
Miastenia gravis de presentación bulbar con timoma. Reporte de Caso y Revisión de Literatura
Resumen
La miastenia gravis (MG) es un desorden de la trasmisión neuromuscular producto de la disminución autoinmune mediada por anticuerpos de los receptores postsinápticos de acetilcolina de la placa motora.
Importancia: Alrededor del 15% presentan síntomas bulbares, caracterizado por debilidad y/o fatiga muscular fluctuante predominantemente en los músculos inervados por los nervios craneales como lo son los músculos oculares y bulbares. Se presenta un caso de un adulto joven masculino con miastenia gravis, con timoma y presentación bulbar, con cuadro clínico caracterizado por dificultad para la masticación, dificultad para deglutir líquidos más que los sólidos (disfagia) y menor tono vocal, voz nasal. La miastenia bulbar, es una forma de MG muy difícil de diagnosticar, porque la afección de los músculos de la deglución, de la cara, de cuello, y respiratorios, se presenta disfagia, disartria, animia, anorexia, debilidad cervical y dificultad respiratoria.
Palabras clave: Miastenia Gravis; autoinmune; bulbar; debilidad muscular; timoma; timectomía.
Abstract
Myasthenia gravis (MG) is a disorder of neuromuscular transmission due to the autoimmune decrease mediated by post-synaptic acetylcholine receptors of the motor plate. Importance: Around 15% have bulbar symptoms, characterized by weakness and / or fluctuating muscle fatigue predominantly in the muscles innervated by the cranial nerves such as the ocular and bulbar muscles. We present a case of a young male adult with myasthenia gravis with thymoma and bulbar presentation with a clinical picture characterized by difficulty in chewing, difficulty swallowing liquids more than solids (dysphagia) and lower vocal tone, nasal voice, dysarthria. Myasthenia Bulbar, is a form of MG very difficult to diagnose, because, the condition of swallowing, face, neck, and respiratory muscles, dysphagia, dysarthria, animia, anorexia, cervical weakness and respiratory distress.
Keyword: Myasthenia Gravis; autoimmune; bulbar muscular weakness; thymoma; timectomy.
Introducción
La miastenia gravis (MG) fue descrita por primera vez por el médico inglés Sir Thomas Willis en 1672, cuya denominación proviene del griego mio músculo, astenia debilidad y gravis intensa, es un trastorno neuromuscular autoinmune caracterizado por debilidad y fatigabilidad de los músculos esqueléticos localizada o generalizada, a predominio proximal y de curso fluctuante debido a la disfunción de la unión neuromuscular (1).
Los síntomas provienen del bloqueo post-sináptico de la transmisión neuromuscular por anticuerpos contra los receptores de acetilcolina y otras proteínas de la membrana postsináptica. No obstante, se plantea su origen autoinmune en la demostración de los anticuerpos antireceptor de ACh que son los responsables del bloqueo de los receptores de acetilcolina (ACh) de la unión neuromuscular debido a la presencia de anticuerpos antireceptor de ACh (2,3).
Debido a lo inusual, puede pasar inadvertida en sus manifestaciones clínicas iniciales, lo que retarda el diagnóstico. El 20% de todas las MG debutan en las dos primeras décadas de la vida, de ellos sólo el 4% antes de los 10 años (3,4).
La MG más frecuente es la generalizada en el 80% de los casos, mientras que la localizada se presenta apenas en el 20%, aunque hasta el 33% de las formas generalizadas comienzan afectando primero a los músculos extraoculares y 10% debutan como crisis miasténicas (5).
La miastenia gravis se manifiesta por fatiga muscular precoz con progresión a la parálisis durante estados de contracción muscular interactivos (movimientos) o sostenidos (posturas). En la mayoría de los casos, comienza con afectación de los músculos oculares, con diplopía o ptosis palpebral. La debilidad puede permanecer confinada a los músculos oculares o comprometer la musculatura que controla la masticación, deglución y articulación de la palabra, los llamados músculos bulbares; en estos casos puede ser difícil el diagnóstico. Cuando la debilidad compromete a la musculatura de las extremidades el diagnóstico es más evidente (4,5).
Casi todos los pacientes con MG tienen anticuerpos contra AchR. La hiperplasia del timo o el timoma se ha visto en el 75% de los pacientes con MG. Por lo tanto, se sospecha que el timo es el sitio de producción de anticuerpos, pero el estímulo que inicia el proceso autoinmune es desconocido. Sin embargo, en la actualidad todavía no se ha entendido completamente la relación entre la glándula del timo y la miastenia gravis, se cree que posiblemente la glándula del timo genere instrucciones incorrectas sobre la producción de anticuerpos receptores de acetilcolina, y se crea así el ambiente perfecto para un trastorno en la transmisión neuromuscular (6,7).
Existen tres formas clínicas de MG, ocular, bulbar y generalizada. La MG ocular es la más frecuente, los pacientes refieren diplopía y ptosis palpebral de rápida progresión, que se incrementa durante el día, bilateral y asimétrica, sin afección pupilar. Solo en el 25% de los casos la enfermedad se generaliza en el curso de dos años y es frecuente que los anticuerpos AchR estén positivos (1-8).
El diagnóstico de MG en la mayoría de los pacientes demora entre tres y nueve meses desde el comienzo de la sintomatología. Esto es debido a que no se identifica la enfermedad y en muchos casos la sintomatología es tomada muy a la ligera. Los primeros pasos para diagnosticar la miastenia gravis incluyen una revisión del historial médico del paciente, el estudio neurofisiológico, el estudio serológico y los test farmacológicos (Test del Tensilon). Prueba de Estimulación repetitiva de nervio periférico (ERN) Prueba de Neostigmina. La tomografía computarizada (CT) o la resonancia magnética (RM) se pueden utilizar para identificar una glándula del timo anormal o la presencia de un timoma como resultó en nuestro caso [10].
El tratamiento de la miastenia gravis se aborda desde dos perspectivas, fármacos anticolinesterásicos para el control de los síntomas e inmunosupresores como tratamiento de base y cirugía. La timectomía es el tratamiento predilecto para los pacientes con miastenia que tienen un timoma. A pesar del avance en los tratamientos, la timectomía sigue siendo una parte integral en el abordaje del paciente con miastenia gravis.
El objetivo de esta investigación es presentar el caso de un paciente con miastenia gravis con un timoma y presentación bulbar, de difícil diagnóstico, informar y dar a conocer su sintomatología de tan inusual presentación, que puede dar lugar a confusión diagnóstica y retardar su tratamiento. Así como también la revisión y actualización de la literatura sobre el tema.
Reporte de Caso Clínico
Paciente masculino de 24 años de edad, con 2 meses desde el inicio de la enfermedad, con cuadro clínico caracterizado por caída de los párpados bilateral (ptosis palpebral), visión doble (diplopía), decaimiento ocasional que exacerba con la actividad física de la musculatura facial y del cuello, dificultad para la masticación, dificultad para deglutir líquidos más que los sólidos (disfagia) y menor tono vocal, voz nasal, disartría , además pérdida de la fuerza muscular en miembros superiores e inferiores.
Después de un mes de iniciada la enfermedad, se agregan al cuadro clínico agotamiento y cansancio fácil, dificultad para respirar a medianos esfuerzos, mayor disfonía y disartría.
Debido a la sintomatología neurológica y que durante este tiempo persistía la visión doble, con episodios de debilidad, que se iniciaban en los músculos de la masticación, continuaban con disfagia, disartría y dificultad respiratoria, se ordena valoración con neurocirugía donde se solicita Resonancia Magnética Cerebral con fase vascular y Gadolinio la cual fue reportada dentro de límites normales, por lo que le indican prednisona a 25 mg diarios.
En el examen neurológico especializado presenta moderada dificultad respiratoria que incluía músculos accesorios, salivación excesiva, cabeza flexionada hacia delante, pérdida del tono muscular del cuello y de los músculos para la masticación, orientado, con lenguaje y memoria sin alteraciones, discreta ptosis palpebral bilateral, nivel proximal en miembros superiores e inferiores, con trofismo disminuido, hipotonía, coordinación normal, paresia facial periférica bilateral, dificultad para la deglución, voz de baja intensidad, resto de nervios craneales y sensibilidad táctil y dolorosa normales, hiperreflexia.
Ante la presunción diagnóstica de un cuadro de miastenia gravis grado II subgrupo B de acuerdo a la clasificación clínica de Ossermann y Genkisen. Se instauró inmediatamente tratamiento con piridostigmina 60 mg cada 4 horas y prednisona a 25 mg/día, se solicitaron los exámenes complementarios, estudios de imágenes radiografía y resonancia magnética de tórax.
Obteniéndose mejoría el cuadro clínico pero persiste la debilidad de los músculos orofaríngeos (masticación y deglución).
Niega antecedentes personales o familiares de importancia.
Exámenes Complementarios: Hematología, perfil bioquímico, todos con valores dentro de límites normales. Pruebas Inmunológicas: ANA, ANCA, Anti DNA negativos. C3 y C4: Normal. IgE, IgG, IgA e IgM: Normal. Pruebas Tiroidea T3, T4 y TSH Normales. VDRL reactivo 32 diluciones y HIV negativo. Tóxicos en orina negativos. Musculo liso Inmunoflorescencia Negativo
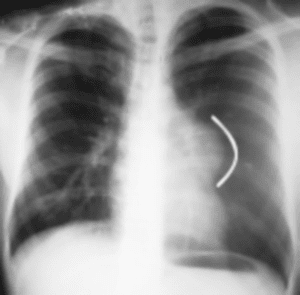
En la radiografía de tórax, se aprecia imagen densa, homogénea, redondeada, bien definida en región mediastinal, que comprende posiblemente al mediastino anterior (Figura. 1).
En la Resonancia Magnética de tórax, en planos y secuencias convencionales en equipo GE EchoSpeed 1.5 T se evidencia lesión de ocupación de espacio localizada a nivel del mediastino anterosuperior redondeada, de contornos bien definidos discretamente heterogéneo que mide 4.3 x 5.5 x 5.1 cm, con mínimo realce periférico, que luego de la administración de contraste endovenoso (Gadolinio) con comportamiento predominantemente hiperintenso en secuencias T2 e hipointenso en secuencias T1 cambios que plantean con los antecedentes y la clínica neuromuscular del paciente compatible con miastenia gravis y la presencia de un Timoma (Figura.2).
Ante la presencia de glándula tímica (Timoma) y del diagnóstico de miastenia gravis y en tratamiento con fármacos anticolinesterásicos para el control de los síntomas. Se remite a la Unidad de Cirugía de Tórax, donde previa valoración se decide conducta quirúrgica, pero previamente a esta se le practica plasmaféresis.
Se lleva a cabo la timectomía a través de esternotomia mediana, se evidencia la presencia de glándula Timica (Timoma), se practica exéresis de este junto a toda la grasa mediastinal, de las pleuras mediastinales. Se deja una sonda torácica en cada cavidad pleural para drenaje y restitución de la presión negativa pleural.
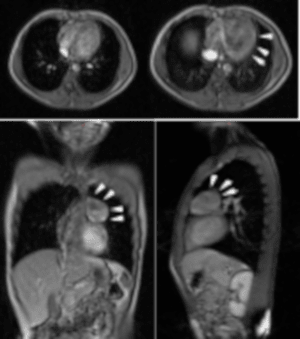
Figura 2. Resonancia Magnética de Tórax. Se evidencio lesión de ocupación de espacio localizada a nivel del mediastino anterosuperior redondeada, de contornos bien definidos discretamente heterogéneo que mide 4.3 x 5.5 x 5.1 cm, con mínimo realce periférico, que luego de la administración de contraste endovenoso (Gadolinio) con comportamiento predominantemente hiperintenso en secuencias T2 e hipointenso en secuencias T1 (flechas blancas), compatible con timoma.
Al final de la cirugía el paciente presento una adecuada autonomía respiratoria, tenía una adecuada respuesta a los comandos neurológicos, sin embargo, se mantiene bajo intubación y es trasladado a la Unidad de Cuidados Intensivos (UCI), para manejo ventilatorio del paciente para su recuperación posoperatoria anestésica. No se utilizaron durante el transoperatorio anticolinesterasicos debido a su buena autonomía respiratoria, pero sobre todo a que en estudios previos se ha podido observar que el uso de estos agentes podría causar un bloqueo muscular despolarizante.
Su evolución extrahospitalaria siguió siendo tórpida persisten los síntomas localizados neuromuscular de la esfera orofaringea la debilidad de los musculo del cuello, sialorrea excesiva, paresia facial periférica bilateral, dificultad para la deglución, sin embargo mantiene conservada la fuerza muscular de los miembros superiores e inferiores, sin problemas respiratorio por lo que el caso presenta varias características interesantes, tales como la ausencia de mejoría del cuadro clínico de la miastenia grave (MG), ya que persisten los síntomas bulbares. La literatura reporta que alrededor del 15% de los pacientes presentan estos síntomas bulbares, como nuestro paciente.
Los síntomas incluyen disartria, disfagia y fatiga al masticar. Menos del 5% se presentan con debilidad en las extremidades (9-12). Por lo que estamos antes un paciente con una miastenia gravis de presentación bulbar con timoma.
La debilidad puede permanecer confinada a los músculos oculares, o comprometer la musculatura que controla la masticación, deglución y articulación de la palabra, llamados músculos bulbares o glosofaríngeos; en estos casos se hace difícil el diagnóstico.
El estudio anatomopatológico definitivo reportó: Timoma tipo B2, según la clasificación de la Organización Mundial de la Salud (OMS) (Figura.4).
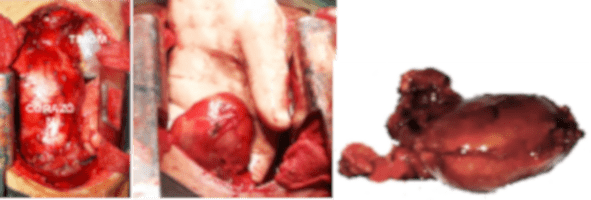
Los timomas se clasifican en cinco subtipos histopatológicos (OMS). El timoma está formado en su mayoría por linfocitos T y células epiteliales. La clasificación de la OMS se basa en la morfología de estas células epiteliales y la cantidad de linfocitos T asociados, que es un indicador de la función biológica de las células del timoma. Los timomas tipos A, AB, B1, B2 y B3 muestran una cierta cantidad de linfocitos T inmaduros, los carcinomas tímicos no tienen un número mesurable de linfocitos T inmaduros y, por lo tanto, no se diferencian. El timoma cortical (tipo B2) se asocia con miastenia gravis en el 50% de los casos, al igual que el paciente descrito, mientras que el timoma medular (tipo A) rara vez se asocia con la miastenia gravis (7,8).
Actualmente seis meses después del posoperatorio, el paciente luce en buenas condiciones generales en franca mejoría de la sintomatología de los músculos bulbares, sin la administración de anticolinesterásicos ni esteroides.
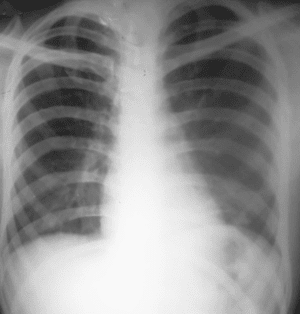
Figura 6. Radiografía Tele de tórax posoperatoria. Dentro de límites normales con buena disposición de las estructuras, se aprecia cuerpo extraño metálico (alambre quirúrgico).
Discusión
La miastenia gravis (MG), en su forma congénita o adquirida, es una enfermedad autoinmune causada por la presencia de autoanticuerpos dirigidos al receptor nicotínico de acetilcolina, que provocan una transmisión insuficiente del impulso nervioso hacia las fibras de músculo estriado, expresada mediante paresia progresiva.
La miastenia gravis es un desorden crónico autoinmune en el que existen anticuerpos séricos que al unirse a los receptores de acetilcolina nicotínicos de la membrana muscular de la placa motora alteran la transmisión neuromuscular. Estos anticuerpos son del tipo IgG y pueden ser detectados en 80-90% de pacientes con MG generalizada y en 50-70% en pacientes con MG grave ocular. Por otro lado, se denomina MG seronegativa a la relacionada a factores humorales y en 40% de los pacientes con esta MG seronegativa pueden encontrarse anticuerpos IgG específicos contra la cinasa específica muscular (muscle specifi c kinase, MuSK, en inglés), lo cual no ocurre en pacientes con MG seropositiva (2).
Se presenta en todas las edades de escasas o poca incidencia en las edades pediátricas en un 15% de los casos (1-4).
En la MG generalizada los síntomas principales fluctúan entre debilidad muscular y fatiga muscular, se inicia con síntomas oculares, que luego de un periodo de tiempo semanas o meses pueden llegar a afectarse la musculatura de los miembros superiores e inferiores sobre todo en su segmento proximal; además la debilidad de la musculatura de los músculos del cuello y los músculos bulbares que pueden condicionar al paciente a la necesidad de soporte ventilatorio; donde los cuadros infecciosos pueden precipitar o exacerbar estos últimos síntomas. Más del 50% de los pacientes presentan síntomas oculares: ptosis y/o diplopía. Alrededor de 15% presentan síntomas bulbares, como el paciente previamente descrito. Los síntomas incluyen disartria, disfagia y fatiga al masticar. Menos del 5% se presentan con debilidad en las extremidades (12).
El caso reportado tuvo una presentación típica de la MG ocular por la forma como se inició la enfermedad con visión doble (diplopía), pero se fue tornando en la de tipo bulbar, debido a que se fueron agregando la debilidad facial y en pocos días se añadió ptosis palpebral y debilidad de los musculo del cuello, dificultad para la masticación y la musculatura bulbar; disartria, hipotonía, disfagia y dificultad respiratoria; síntomas que se acentuaban al transcurrir los días y que mejoraban transitoriamente con el descanso. Estas manifestaciones reflejan la fatigabilidad de los músculos bulbares que se soslayaron en las primeras atenciones (12,13).
Esta forma de presentación, fue descrita por primera vez en 1868 por Hérard como una variante de “parálisis del glosofaríngeo”, afecta a los músculos de la masticación acompañada de una marcada dificultad para tragar líquidos con regurgitación nasal, disfagia mayor a los líquidos que a los sólidos, y disartria, sintomatología que refirió el paciente. Esta forma de MG es difícil de diagnosticar y se complica con neumonía por broncoaspiracion alimenticia. Nuestro paciente estuvo dos meses sin el diagnóstico correcto, y fueron confundidos sus síntomas con hiperreactividad bronquial (asma), tal y como se describe en la literatura. Otros hallazgos frecuentes de la MG bulbar, son la pérdida de peso, que se confunde con anorexia primaria como fue el caso del paciente lo que motivo su evaluación por psiquiatría (13).
Cuando el paciente inició su cuadro clínico de enfermedad con afección de los músculos de la cara y después de los músculos bulbares, ya tenía una condición seria, por lo que la severidad de su cuadro fue confundida al no tomar en cuenta toda la información, no se reconoció la fatigabilidad debido a que pudo haber existido la posibilidad de que su mejoría parcial se debió al efecto inducido por los corticoides, hasta que se comprometieron los músculos de la masticación y de la deglución (20-30% de los pacientes). Es importante el diagnóstico basado en una buena historia y un buen examen físico, sobre todo en la esfera neurológica.
El paciente según la clasificación clínica de la de la Fundación Americana de Miastenia Gravis (MGFA), alcanzo la Clase III, de severidad. Siempre es recomendable la clasificación de la enfermedad para iniciar las estrategias terapéuticas (11-14).
También es necesario utilizar los métodos más fiables de laboratorio como la confirmación con pruebas serológicos: anticuerpos (anticuerpo contra receptor de acetilcolina y anticuerpos contra el receptor de tirosina cinasa musculo especifico) y pruebas electrofisiológicas (prueba de estimulación nerviosa repetitiva y el electromiograma de fibra única) (12).
La exploración imagenológica del mediastino debe realizarse siempre en el paciente con miastenia gravis, en especial con tomografía computada y/o resonancia magnética torácica, técnicas que nos permiten revelar la presencia de un tumor tímico. La presencia de un tumor en la glándula tímica en el paciente miasténico, se presenta en el 15-20% de los casos y esta circunstancia cambia el pronóstico de la enfermedad, como sucedió en el caso que estamos reportando.
No existe un régimen terapéutico uniforme de elección para los pacientes con MG, de manera que el tratamiento debe ser individualizado en razón de cada paciente y basarse en las características clínicas, entre las cuales se incluye la distribución, duración y gravedad de la debilidad. Asimismo, se debe tener en cuenta la presencia de un timoma.
En cuanto al tratamiento este tiene cuatro componentes: 1. Tratamiento sintomático (agentes anticolinesterásicos), 2. tratamientos crónicos inmunomoduladores (glucocorticoides e inmunosupresores), 3. tratamientos inmunomodulares rápidos (plasmaféresis e inmunoglobulinas intravenosas), y 4. tratamiento quirúrgico (timectomía).
La cirugía bien sea convencional (esternotomia mediana) o torácica mínimamente invasiva (vídeo-asistida), es la modalidad principal para el tratamiento de los pacientes con miastenia gravis y timoma. La cirugía permite evaluar el componente histopatológico exacto y la estadificación, y es la modalidad de tratamiento de primera línea de elección en la mayoría de los casos. Por ello se recomienda la resección quirúrgica inmediata y completa para tumores resecables. La eliminación radical del timoma es curativa para los tumores tímicos en la mayoría de los casos, aunque los pacientes siguen padeciendo miastenia gravis después de la cirugía. Por lo tanto, es necesario el tratamiento farmacológico y el seguimiento continuo después de la cirugía (15-18).
Por ultimo recientemente se encuentran en investigación procedimientos terapéuticos dirigidos a interferir el mecanismo inmunológico que se desencadena en MG, eliminando la respuesta autoinmune en contra del R-ACh. Esta línea de investigación se basa en (a) Estudios sobre linfocitos B, para la interrupción de la acción de la célula B disminuyendo o anulando la formación de anticuerpos del R-ACh; (b) estudios sobre la célula T, con anticuerpos monoclonales anti-CD4, lo cual reduce la respuesta de este grupo celular en la patogenia de MGJ; (c) la utilización de toxina anti-IL 2 que genera el complejo toxina- IL2 que actúa en el receptor de IL2 del linfocito T disminuyendo su estimulación; (d) globulina antilinfocitaria que generaría una inhibición del linfocito T CD4 y estimulación del linfocito T supresor, lo cual llevaría a una supresión o disfunción en la síntesis de anticuerpos. Todos estos estudios se encuentran en etapa experimental y no han sido evaluados en pacientes (19).
Conclusión
La miastenia gravis tiene una conexión muy intrínseca con los timomas desde el punto de vista de su fisiopatológia, por lo que es un cuadro clínico a menudo olvidado en la práctica médica habitual, su diagnóstico se hace un tanto difícil cuando los pacientes consultan por síntomas de debilidad muscular fluctuante, en especial si presentan afectación ocular, como diplopía o ptosis palpebral, o síntomas bulbares, como disfagia o problemas de masticación o fonación.
La Miastenia Bulbar, es una forma de MG muy difícil de diagnosticar, porque, la afección de los músculos de la deglución, de la cara, del cuello, y respiratorios, se presenta disfagia, disartria, animia, anorexia, debilidad cervical y dificultad respiratoria. Estos signos, explorados en forma aislada, desvían la atención y pueden confundirse con otros cuadros médicos complejos.
Por todo lo expuesto su tratamiento es multimodal e incluye tratamiento farmacológico y quirúrgico. La extirpación del timo permite en muchos casos un mejor control de la enfermedad y siempre que sean resecables debe ser parte del tratamiento inicial.
La enfermedad miasténica es un cuadro clínico que debemos conocer, dado que con su detección y tratamiento precoz se consigue generalmente una gran mejoría física y funcional que hace que la mayoría de los pacientes puedan recuperar sus actividades y su modo de vida habitual.
Conflictos de intereses
El autor no tiene ningún conflicto de interés en el desarrollo de la investigación.
Referencias bibliográficas
- Alvarado-Merino, Rosa Y, Espíritu, Elizabeth R, Juárez, Tania, Cok, Jaime, Ferrufino, María C, Samalvides, Susan K, Espinoza, Iván O, Vila, Judith R, & Guillén-Pinto, Daniel. Miastenia gravis de tipo bulbar en niños: un caso de difícil diagnóstico. Rev Neuropsiquiatr [Internet]. 2017 Abr [citado 2019 may 09]; 80(2): 144-150. Disponible en: http://dx.doi.org/https://doi.org/10.20453/rnp.v80i2.3094.
- Briones-Vega, CG. Viruez-Soto, JA. Vallejo-Narváez, CM. Tórrez-Morales, Froilán. Briones-Garduño, JC. Díaz de León-Ponce, MA. Plasmaféresis en miastenia grave y embarazo. Revista de la asociación Mexicana de Medicina Critica y Terapia Intensiva [Internet]. 2015 [citado 2019 may 19]; 29(1):46-49. Disponible en: http://www.medigraphic.com/medicinacritica.
- Breiner A, Widdifield J, Katzberg HD, Barnett C, Bril V. Epidemiology of myasthenia gravis in Ontario, Canada. Neuromusular disorders [Internet]. 2016 [citado 2019 may 22]; 26(1):41–6. Disponible en: doi: http://dx.doi.org/10.1016/j.nmd.2015.10.009.
- Rybojad B, Lesiuk W, Fijałkowska-Nestorowicz A, Rybojad P, Sawicki M, Lesiuk L. Management of myasthenic crisis in a child. Anestezjol Intens Ter [Internet]. 2013 [citado 2019 jun 1];45(2):82
- Gilhus NE, Verschuuren JJ. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol [Internet]. 2015 [citado 2019 jun 1];14(10):1023–36. Disponible en: http:// dx.doi.org/10.1016/S1474-4422(15)00145-3.
- Berrih-Aknin S, Frenkian-Cuvelier M, Eymard B.Diagnostic and clinical classification of autoimmunemyasthenia gravis. J Autoimmun [Internet].2014[citado 2019 jun 10]; 48-49:143-8 . http://dx.doi: 10.1016/j.jaut.2014.01.003
- Melzer N, Ruck T, Fuhr P, Gold R, Hohlfeld R. Clinical features , pathogenesis , and treatment of myasthenia gravis : a supplement to the Guidelines of the German Neurological Society. J Neurol [Internet]. 2016 [citado 2019 jun 19];263(8):1473- 1494. Disponible en: doi: http://dx.doi: 10.1007/s00415-016-8045-z.
- Zisimopoulou P, Evangelakou P, Tzartos J, Lazaridis K, Zouvelou V, Saruhan-direskeneli G, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun [Internet]. 2014 [citado 2019 jun 25];52:139–45. Disponible en: http://dx.doi.org/10.1016/j.jaut.2013.12.004.
- Arosemena Coronel Marilyn. Miastenia Gravis de Presentación Bulbar: Reporte de Caso y Revisión de Literatura. Rev. Ecuat. Neurol [Internet].2017[citado 2019 jul 25];. 26( 3):289-291. Disponible en: http://revecuatneurol.com/
- Gómez Sergio, Álvarez Yelitza, Puerto Jorge Andrés. Miastenia Gravis: una visión actual de la enfermedadMyasthenia Gravis: a current vision of disease. Medicas UIS [Internet]. 2013 Dec [citado 2019 ago 09] ; 26( 3 ): 13-22. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S012103192013000300002&lng=en.
- Ha JC, Richman DP. Biochimica et Biophysica Acta Myasthenia gravis and related disorders : Pathology and molecular pathogenesis. BBA – Mol Basis Dis [Internet]. 2015 [citado 2019 jul 5];1852(4):651–7. Disponible en: Disponible en: http://dx.doi.org/10.1016/j. bbadis.2014.11.022.
- Jayawant S, Parr J, Vincent A. Autoimmune myasthenia gravis. Handb Clin Neurol [Internet]. 2013 [citado 2019 jul 19]; 113: 1465-8. Disponible en: http://DX.DOI: 10.1016/B978-0-444-59565-2.00015-0.
- Khadilkar S V, Chaudhari CR, Patil TR, Desai ND, Jagiasi K, Bhutada AG. Once myasthenic, always myasthenic? observations on the behavior and prognosis of myasthenia gravis in a cohort of 100 patients. Neurol India [Internet]. 2014 Sep-Oct [citado 2019 jul 19]; 62(5):492–7. Disponible en: http:// DOI: 10.4103/0028-3886.144438.
- Oger J, Frykman H. An update on laboratory diagnosis in myasthenia gravis.Clin Chim Acta [Internet]. 2015 [citado 2019 jul 19];449:43-8. Disponible en: http://dx.doi: 10.1016/j.cca.2015.07.030.
- Rückert JC, Sobel HK, Gohring S, Einhaupl KM, Müller JM. Matched-pair comparison of three different approaches for thymectomy in myasthenia gravis. Surg Endosc [Internet]. 2003 [citado 2019 jul 20];17:711-5. Disponible en: http:// DOI: 10.1007/s00464-002-9162-6
- Jans B Jaime, González L Roberto. Resultados de la cirugía torácica mínimamente invasiva (vídeo-asistida) en el tratamiento de la Miastenia Gravis. Rev Chil Cir [Internet]. 2013 [citado 2019 jul 22]; 65( 1 ): 64-72. Disponible en: http://dx.doi.org/10.4067/S0718-40262013000100013.
- Gilhus NE, Verschuuren JJ. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol [Internet]. 2015 [citado 2019 jul 22];14(10):1023–36. . Disponible en: http:// dx.doi.org/10.1016/S1474-4422(15)00145-3
- Schiavi A, Papangelou A, Mirski M. Preoperative Preparation of the Surgical Patient with Neurologic Disease. Med Clin NAm [Internet]. 2009 [citado 2019 jul 26]; 93:1123-1130. Disponible en: http: // DOI: 10.1016/j.anclin.2009.09.011.
- Meriggioli MN, Sanders DB. Autoinmune myasthenia gravis: emerging clinical and biological heterogenetity. Lancet Neurol [Internet]. 2009 [citado 2019 jul 28]; 8(5):475-90. Disponible en: http: // DOI: 10.1016/S1474-4422(09)70063-8.

Autores
Dr. Juan C. Araujo C.
Cirujano General y Cirujano de Tórax.
Facultad de Medicina.
Escuela de Medicina Universidad del Zulia
- Dr. Juan C. Araujo C.#molongui-disabled-link
- Dr. Juan C. Araujo C.#molongui-disabled-link
- Dr. Juan C. Araujo C.#molongui-disabled-link
- Dr. Juan C. Araujo C.#molongui-disabled-link