
Variante genética y enfermedad: la respuesta está en la clínica y en la familia
Estamos viviendo una eclosión de información sobre variantes genéticas asociadas con las enfermedades hereditarias gracias a la secuenciación masiva y al abaratamiento de sus costes. Ser portador de una variante puede no tener implicaciones patológicas, y continúa siendo fundamental vincular los cambios encontrados en el genoma con la clínica. Presentamos un caso que da buena cuenta de ello.
Se trata de una mujer de 47 años remitida a consulta de nefrología desde atención primaria para realizar un seguimiento por poliquistosis renal autosómica dominante (PQRAD). Su hijo, de 17 años, ha sido incidentalmente diagnosticado de PQRAD, según los criterios de Pei et al.1 en el contexto de dolor abdominal. Se llevó a cabo un estudio genético en el hijo para confirmar el diagnóstico, debido a la ausencia de antecedentes familiares de la enfermedad y a la existencia de estudios ecográficos normales en todos los familiares de primer grado. Se identificaron tres variantes en heterocigosis en el gen PKD1, no descritas hasta el momento, y que se clasifican de significado incierto. Sin embargo, los análisis in silico las califican como patogénicas (tabla 1). La paciente está asintomática, PA 110/60 mmHg, creatinina sérica 0,68 mg/dL, sin alteraciones urinarias y la ecografía renal es completamente normal. Se realiza un estudio genético, que identifica en heterocigosis las variantes c.3999C> G y c.5068G> A en el gen PKD1, previamente localizadas en su hijo. En esta situación, se repite una nueva ecografía renal, que vuelve a no presentar hallazgos patológicos, por lo que se realiza una resonancia abdominal en la que tampoco se encuentra ninguna anormalidad (fig. 1).
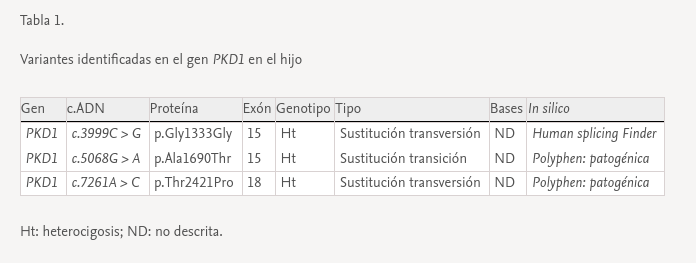
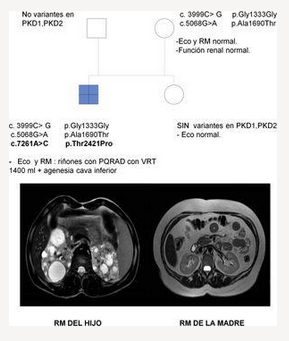
Se efectuaron estudios genéticos al padre y a la hermana del caso índice en el contexto de estudio de segregación y no se identificó variante alguna (fig. 1).
El hijo ha heredado las dos variantes en el gen PKD1 vía materna, y muestra otra (c.7261A> C), que se sospecha que podría ser de novo. Esta no está descrita y produce, a nivel proteico, un cambio de una treonina por una prolina en la posición 2421, el análisis in silico le asigna un carácter patogénico.
A pesar de los avances en genética, el diagnóstico de PQRAD continúa siendo clínico al día de hoy. Los estudios genéticos permiten esclarecer los diagnósticos, pero en algunas ocasiones, pueden llevar a confusión. La interpretación de los estudios genéticos no es sencilla y requiere de entrenamiento. Cada vez más, los clínicos tendrán que familiarizarse con la nomenclatura específica de los mismos y con la búsqueda de las variantes en las bases de datos internacionales habilitadas para compartir el conocimiento sobre estas. La genética no debe separarse de la clínica y del estudio de las familias porque esto podría llevar a una inadecuada valoración de los resultados, como ejemplifica este caso clínico. Se debe informar en las consultas de las limitaciones que presentan los estudios genéticos y qué es lo que se puede esperar de ellos. Ser portador de una o varias variantes genéticas en un gen determinado no es sinónimo de enfermedad. Por otro lado, en la PQRAD existe una gran variabilidad intrafamiliar, por lo que, a pesar de compartir la variante genética, la evolución de la patología puede ser muy distinta de unos individuos a otros por mecanismos que aún se desconocen2,3. Sin embargo, a pesar de estas limitaciones, los estudios genéticos tienen, hasta el día de hoy, una gran vertiente clínica en el manejo de la PQRAD y son fundamentales para poder adelantarse al diagnóstico de la enfermedad. Aún existe controversia sobre cuándo es el momento más adecuado para realizar el despistaje en los hijos de pacientes afectados, ya que no existe todavía un tratamiento curativo, pero es indudable que los pacientes portadores de enfermedades hereditarias tienen derecho a un adecuado asesoramiento genético4,5. El grupo de estudio de la enfermedad poliquística autosómica dominante (GEEPAD) ha demostrado que, en el 60% de las ocasiones, el diagnóstico de PQRAD se realiza a una edad media de 34 años y tras haber tenido ya el primer hijo6.
La única manera de adelantarse al diagnóstico de la PQRAD es filiar adecuadamente a todas las familias y efectuar estudios genéticos. La realización de árboles genealógicos, estudios genéticos secuenciales y de segregación, cuando sea necesario, debe ser también abordada por los nefrólogos. El paso de un abordaje individual a uno familiar es fundamental en el buen manejo de esta y otras entidades renales hereditarias.
Este caso clínico pone en relieve la necesidad de aunar la experiencia clínica y la genética. Este desafío va a estar cada día más presente en la labor asistencial diaria y es un cambio al que hay que adaptarse.
Bibliografía
Autores
- Servicio de Nefrología, Hospital Universitario Clínico San Cecilio. Granada, España
- Servicio de Nefrología, Hospital Universitario Virgen de las Nieves. Granada, España

Autores
Revista Nefrología
Nefrología es la publicación oficial de la Sociedad Española de Nefrología. La revista publica artículos sobre investigación básica o clínica relacionada con la nefrología, la hipertensión arterial, la diálisis y el trasplante de riñón.
- Revista Nefrología#molongui-disabled-link
- Revista Nefrología#molongui-disabled-link
- Revista Nefrología#molongui-disabled-link
- Revista Nefrología#molongui-disabled-link