
Lupus eritematoso sistémico
Aún en la naciente era de la biología molecular y a pesar del avance enorme en el desarrollo de la inmunología como ciencia, la etiopatogenia del lupus eritematoso sistémico (LES) continua siendo un enigma. Los modelos murinos representan una aproximación muy útil pero parcial al problema humano y la heterogeneidad clínica del cuadro complica aún más las posibilidades de definir mecanismos causales específicos.
Sin embargo, es evidente que los últimos cinco años han representado un avance importante en nuestro entendimiento del proceso continuo que va desde la ruptura de los finos mecanismos que controlan el reconocimiento de lo propio hasta el daño tisular multisistémico.
En los momentos actuales podemos identificar y ubicar genes de susceptibilidad con las nuevas técnicas de oligotipeaje usando la amplificación de genes mediante la reacción en cadena de la polimerasa (PCR); estamos en capacidad de investigar el origen de los autoanticuerpos examinando el uso de genes de inmunoglobulina (lg) a nivel de línea germinal o por el proceso de rearreglos de sus regiones variables como supone una respuesta dirigida por antígenos. Podemos también identificar los autoantígenos que posiblemente estimulan la producción de estos auto anticuerpos Y entendemos mejor la cascada de eventos bioquímicos subcelulares que llevan a la activación de las diversas células inmunocompetentes.
Es obvio que conocemos más del proceso. La pregunta es si ahora lo entendemos mejor. Con todas estas importantes piezas del rompecabezas en la mano el reto es como organizarlas en un modelo coherente, que de respuesta a muchas interrogantes, entre ellas la identificación del evento crítico inicial o la falla fundamental responsable de la ruptura de la tolerancia inmunológica para un número considerable de autoantígenos en el LES.
A continuación pasaremos a comentar brevemente algunos de los avances más recientes relevantes a la etiopatogenia del LES, haciendo énfasis en resultados publicados en los últimos tres años y en nuestros más recientes experimentos.
Alteraciones en el proceso de tolerancia inmunológica
Uno de los hallazgos más relevantes es el defecto en la expresión de la proteína fas, molécula clave en el proceso fisiológico de apotosis o muerte celular programada,1 en el modelo murino MRL/lpr/lpr. Este hallazgo explicaría la linfoproliferación anormal de linfocitos CD4-/CD8- observada en estos ratones. El defecto se debe a la inserción del retrotransposón ETn en el segundo intrón del gen fas, alterando el empalme1 de los exones del gen, con la consiguiente expresión defectuosa de la proteína.2
Un retrotransposón es una secuencia retroviral que se transcribe en forma reversa en el ADN genómico de cualquier célula. Su presencia en este modelo murino de lupus refuerza las evidencias del posible papel de los retrovirus en la etiopatogenia de algunas enfermedades autoinmunes. En humanos se estima que los retrotransposones representan un 5 % del ADN genómico normal. Otros ejemplos del papel de estas secuencias retrovirales en enfermedad humana son las inserciones en el gen del factor VIII en el cromosoma X responsable de un porcentaje de los casos de hemofilia o en el gen de la distrofina causando la distrofia muscular tipo Duchenne.
Los defectos en la apoptosis ocasionados por esta mutación genética, podrían alterar los proceso de selección negativa intra y extra-tímica indispensables para la eliminación de clonas potencialmente autorreactivas. Sin embargo, tanto en el LES murino como humano hay reportes todavía contradictorios respecto a la presencia de alteraciones en el proceso de apoptosis.
La importancia que estos mecanismos podrían tener en determinar autoinmunidad se ilustra con el modelo transgénico2 para el gen bc/2.3 Al contrario del receptor fas, la proteína codificada por el gen bc/2 inhibe el proceso de apoptosis, pudiendo así interferir con el proceso de tolerización del animal. El transgénico bc/2 muestra un aumento en la sobrevida de los linfocitos D, hipergammaglobulinemia, altos títulos de anticuerpos anti-ADN y manifestaciones autoinmunes sistémicas en cepas de animales que por lo demás serían completamente normales. Hay también reportes contradictorios respecto a la presencia de expresión aumentada del gen bc/2 en el LES humano, siendo éste un campo muy activo de investigación actua1mente (revisado en 4).
Citoquinas y LES
Una de las áreas que ha despertado más interés recientemente es la de si existen imbalances en la respuesta tipo TH1 y TH2 en el LES. La respuesta TH1 se define por la producción de interleukina 2 (IL-2 e interferon τ (IFN-τ) por linfocitos τ cooperadores; la respuesta TH2 por la producción de IL-4, IL-5, IL-6, IL-9 e IL-10.
En enfermedades infecciosas de expresión clínica polar como la lepra, la tuberculosis y la leishmaniasis, la respuesta TH1 se asocia con enfermedad clínica e histológicamente limitada, mientras que la respuesta TH2 se asocia con enfermedad enérgica diseminada.
La reacción injerto contra huésped crónica es un modelo clínico de autoinmunidad. En la respuesta aguda hay un patrón de respuesta tipo TH1, con producción aumentada de IL-2 e IN-τ. Por el contrario, la reacción crónica muestra un patrón TH2 predominante con manifestaciones clínicas similares a las observadas en el LES.5 El papel apaciguador de autoinmunidad de las citoquinas tipo TH1 se evidencia en los reportes que muestran la posibilidad ele transformar una reacción injerto contra huésped aguda en una reacción crónica, mediante la administración de anticuerpos monoclonales anti-IL-2 en animales experimentales (revisado en 6).
Adicionalmente, ciertos modelos murinos de lupus muestran un fuerte patrón ele respuesta TH2. Por ejemplo, el ratón Palmerston-North desarrolla un aumento en la producción de IL-4 e IL-10, que coincidieron la aparición de manifestaciones autoinmunes. En el lupus humano hay mayor transcripción del RNA de IL-6 en linfocitos circulantes. Se especula que un patrón de respuesta TH2 facilitaría la aparición de autoinmunidad, por ser más difícil tolerizar respuestas de linfocitos T inducidas por IL-4, y porque las células T productoras de IL-4, IL-5 e IL-10 se activan más fácilmente aún en ausencia de señales coestimulatorias procedentes de las células presentadoras de antígeno.6 Esto facilitaría la participación de este tipo de células T en la perpetuación de la respuesta autoinmune.
Origen y papel patogénico de los anticuerpos anti-ADN
Estos autoanticuerpos se originan y perfeccionan en su afinidad y avidez mediante estimulación antigénica y no por expansión policlonal de clonas de linfocitos B productoras de Ig. A favor de este concepto está el hecho de que los anticuerpos anti-ADN patogénicos son predominantemente de la clase IgG y muestran múltiples mutaciones somáticas en las regiones hipervariables que son las responsables del reconocimiento del antígeno. Este perfeccionamiento de la capacidad de ligado de los autoanticuerpos necesita el papel regulador de los linfocitos T.
Molecularmente se ha observado una fuerte representación de los aminoácidos de carga básica arginina y lisina en la región hipervariable CDR3 de estas Ig; substituciones de los mismos hacen que los anticuerpos pierdan su reactividad contra el ADN. Hay una gran similitud molecular en los anticuerpos anti-Sm y los anticuerpos anti-ADN, indicando que el estímulo antigénico responsable d e s u aparición es probablemente el mismo.
Los anticuerpos anti-ADN se unen a complejos de histonas y a ADN en el riñón.7 Las histonas son proteínas básicas que forman, junto a una doble vuelta de ADN (de = 200 bp), los nucleosomas. Un reporte reciente mostró niveles elevados circulantes de nucleosomas en el plasma de pacientes con LES,8 indicando que dichos componentes nucleares podrían estimular la producción de estos anticuerpos.
Genes y autoanticuerpos en LES
El estudio de la relación HLA y LES ha mostrado una asociación más fuerte con la producción de determinados autoanticuerpos que con la enfermedad misma. Por ejemplo, la presencia de los anticuerpos anti-Ro está fuertemente asociada a la presencia de los alelos DQA1 *0501 y DQB1*0201 (subtipos previamente designados DQw2.1) y con los alelos DQA1*06 y DQB1*06 (previamente DQw6) en la región HLA-DQ.
La característica molecular que comparten estos antígenos clase II del complejo principal de histocompatibilidad es la presencia del aminoácido glutamina en posición 34 de la región más externa de la cadena DQA1 y de leucina en posición 26 de la cadena DQB1.9
Los anticuerpos anticardiolipina presentes en 25 a 40% de pacientes lúpicos se asocian con la presencia del alelo DQB1*0301 (perteneciente a la especificidad DQw7). Nosotros hemos encontrado una asociación entre estos anticuerpos y el genotipo 18/18 en la cadena lambda de Ig, determinado mediante la técnica de Southern blot.10
Los anticuerpos anti-Sm se asocian a los alelos DQA1*0 102 y DQB1*0602 y los anticuerpos RNP a DQA1*0101 y a los haplotipos DR2 y DR4.
Es llamativo que la predisposición a producir estos distintos autoanticuerpos resida predominantemente en la región HLA-DQ, más que en la región HLA-DR, a pesar de que esta región se ha asociado a susceptibilidad con la enfermedad en su conjunto. Es posible, que una concordancia de alelos en éstas y tal vez otras regiones cercanas del complejo HLA, condicione la expresión de manifestaciones clínicas propias de cada subconjunto clínico. La configuración de alelos en el HLA y otras regiones relevantes para la respuesta inmune determinaría el tipo de manifestación serológica y clínica predominante en el LES.
Alteraciones en la función de linfocitos T
Las tendencias más recientes dan preponderancia a posibles alteraciones en el compartimiento de células T en la patogenia del LES. Así por ejemplo, el tratamiento de ratones lúpicos con anti cuerpos monoclonales antilinfocitos CD4+ o el bloqueo de la interacción IL-2 con sus receptores específicos mejora la sobrevida de estos animales. Además, como se comentó arriba la producción de los autoanticuerpos patogénicos es dependiente de células T, que son las que dirigen el switch a isotipos fijadores de complemento y las hipermutaciones somáticas en sus regiones variables para permitir mayor afinidad y avidez por antoantígenos.
No está claro cuál es el status funcional de los linfocitos T circulantes en el LES. La literatura previa está llena de reportes de respuestas defectuosas a la estimulación con lectinas de planta y de alteraciones en los patrones de producción de citoquinas. Nosotros reportamos recientemente que estas células tenían una respuesta normal o aumentada al ser estimulados por la vía CD3.11 Esta respuesta requiere la presencia de células accesorias o la co-estimulación con ésteres del forbol, que son activadores de la proteína quinasa C. Estos resultados han sido confirmados por otro grupo de investigadores.12 En estudios más recientes observamos que los linfocitos T de pacientes lúpicos activados por la Vía CD4+ responden más vigorosamente a la IL-2.13
No se conoce cuál pueda ser el mecanismo íntimo de esta respuesta. Cada vez se entiende mejor la secuencia de señales subcelulares que van de la estimulación de receptores de membrana a la activación de genes críticos para la respuesta de linfocito T; es posible que algunos defectos en la regulación de estas señales puedan disminuir el umbral para respuesta autoinmunes auto perpetuadas, en situaciones en que los linfocitos T encuentren determinados autoantígenos. En la Figura 1 se muestra un esquema simplificado del conjunto de señales bioquímicas que se disparan luego del ligado del antígeno por el linfocito T.
[su_spacer size=”30″]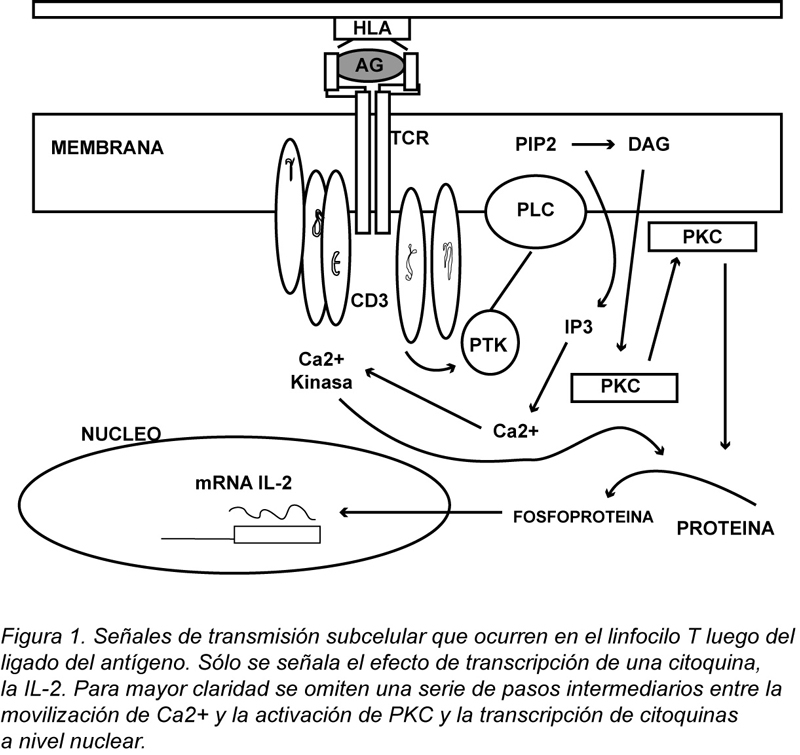 [su_spacer size=”30″]
[su_spacer size=”30″]
Es posible concebir que una subversión en la cascada fisiológica subcelular, por señales anormales derivadas del apagamiento de una quinasa normal o de la aparición de una quinasa anormal codificada por una secuencia retroviral del tipo de retrotransposones arriba mencionados, pueda romper el delicado balance entre tolerancia y autoinmunidad.
Nosotros hemos comenzado a examinar una de las señales intracelulares más tempranas que ocurren luego de la activación por la vía CD3. Esta es la fosforilización de diversos substratos en residuos de tirosina por la acción del sistema enzimático de la proteína tirosina quinasa (PTK). Nuestros resultados preliminares sugieren que los linfocitos T de pacientes lúpicos tienen una respuesta alterada de PTK, como lo indica la fosforilización más débil de proteínas derivadas de células T activadas con el anticuerpo monoclonal OKT3 (Figura 2).
[su_spacer size=”30″]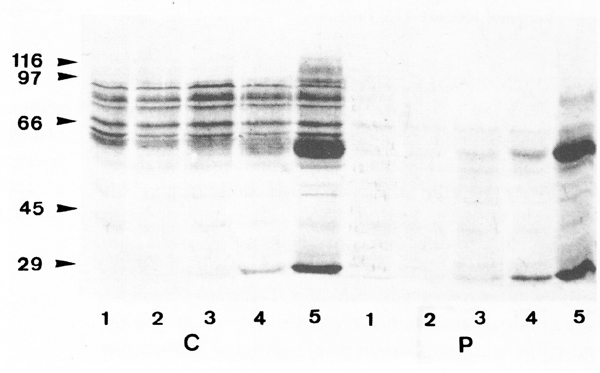 Figura 2. Fosforilización de proteínas en residuos de tirosina en linfocitos T activados por la vía CD3. Se aislaron linfocitos T mediante pasos sucesivos en Ficoll-Hypaque, adherencia a columnas de nailon y gradientes discontinuos de Percoll. 2 x 106 células fueron estimuladas con el anticuerpo monoclonal OKT3, durante 3 minutos, en presencia del anticuerpo de cabra anti-lg de ratón (para inducir “cross-linking ” del complejo CD4+ Las proteínas fosforiladas en tirosina, presentes en los lisados de estas células fueron examinadas por la técnica de Wesren blot. OKT3 0µg carril 1, 0.01 µg carril 2, 1µg carril 3, 10µg carril 4 y 100µg carril 5. C = control sano, P = paciente con LES. A la derecha muestran los pesos moleculares de las bandas de kDa.
Figura 2. Fosforilización de proteínas en residuos de tirosina en linfocitos T activados por la vía CD3. Se aislaron linfocitos T mediante pasos sucesivos en Ficoll-Hypaque, adherencia a columnas de nailon y gradientes discontinuos de Percoll. 2 x 106 células fueron estimuladas con el anticuerpo monoclonal OKT3, durante 3 minutos, en presencia del anticuerpo de cabra anti-lg de ratón (para inducir “cross-linking ” del complejo CD4+ Las proteínas fosforiladas en tirosina, presentes en los lisados de estas células fueron examinadas por la técnica de Wesren blot. OKT3 0µg carril 1, 0.01 µg carril 2, 1µg carril 3, 10µg carril 4 y 100µg carril 5. C = control sano, P = paciente con LES. A la derecha muestran los pesos moleculares de las bandas de kDa.
[su_spacer size=”30″]
Actualmente estamos examinando si ello pueda deberse a alteraciones en la cinética de la respuesta y viendo como se correlaciona esta señal con la transcripción de genes de citoquinas relevantes par la activación y proliferación de linfocitos T■
Referencias
-
Watanabe-Fukunaga R. Brannan CI, Copeland NG, et al. Lymphoproliferation disorder in mice explained by defects in fas antigen that mediates apoptosis. Nature 356:314-317, 1992
-
Wu J, Zhou T, He J, Mountz.ID. The fas gene mutation in Ipr mice is due to insertion of a retroviral transposon. J Exp Med 178:442-451, 1993
-
Strasser A, Whittingham S, Vaux DL, et al. Enforced bc 12 expression in B -lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci USA 88:8661-8665, 1991
-
Isenberg DA, Ehresnstein MR, Longhurst C, Kalsi.I K. The origin, sequence, structure, and consequences of developing anti-DNA antibodies. A human perspective. Arthritis Rheum 37:169-180, 1994
-
De Witt D, van Mechelen M, Zaniun C, et al. Preferential activation of TH2 cells in chronic graft versus host reaction. J Immunol 150:361-372, 1993
-
Mountz JD, Gause WC. Murine models of autoimmune disease and Sjögren’s syndrome. Curr Opin R heum 5:557-569, 1993
-
Termaat RM, Assmann KJM, Dijkman IIBPM, et al. AntiDNA antibodies bind to the glomerulus via two distinct mechanisms. Kidney Int 42:1363-1371, 1992
-
Rumoer P, Steinmann. Endogenous circulating DNA in systemic lupus erythematosus: Occurrence as multimeric complexes bound to histones. J Clin Ivest 86: 69-74, 1990
-
Arnett FC, Reveille. ID. Genetics of systemic lupus erythematosus. Rheum Dis Clin NA 18:865-892, 1992
-
Blasini AM. Polimorfismos en la longitud de los fragmentos de restricción para los genes de la región constante de la cadena lambda de las inmunoglobulinas en el lupus eritematoso sistémico. Tesis MSc. IVIC, 1993.
-
Stekman IL, Blasini AM, León-Ponte M. Baroja ML, Abadí I, Rodríguez MA, Enhanced CD3-mediated T lymphocyte proliferation in patients with systemic lupus eruthematosus. Arthritis Rheum 34:459-467, 1991.
-
Stohl W. Impaired generation of policlonal T cll-mediated cytolyticactivity despite normal polyclonal T cell proliferation in systemic lupus erythematosus. Clin Immunopathol 63:163-172, 1992.
-
Blasini AM, Stekman IL, González F. Tositi ML, Rodríguez MA. T lumprhocytes from patients with systemic lupus erythematosus show increased response to in terleukin-2 after costimulation with OKT3 monoclonal antibody and phorbol esters. Clin Immunopathol 70:66-72, 1994.
