
Histiocitosis de células de langerhans asociado a inmunodeficiencia primaria: a propósito de un caso
Resúmen
Las Histiocitosis son un grupo de enfermedades, caracterizadas por la proliferación de células del sistema fagocitico- mononuclear. Dentro de este grupo se encuentra la histiocitosis de células de langerhans, la cual se caracteriza por la proliferación clonal de células dendríticas, que puede afectar a distintos órganos y sistemas. La incidencia es desconocida siendo más frecuente en la infancia especialmente durante el primer y cuarto año de vida. El diagnóstico definitivo se establece mediante biopsia de la lesión, en las que se observan células de langerhans patológicas (CD1- CD207 positivo). Las Inmunodeficiencias primarias constituyen un grupo de defectos genéticos, que generan alteración en los mecanismos de defensas tanto innatos como adaptativos, que condicionan no solo a susceptibilidad elevada para infecciones por diferentes agentes infecciosos sino también a la alteración de los mecanismos hemostáticos y de vigilancia que evitan el desarrollo de enfermedades autoinflamatorias y neoplásicas.
Palabras Clave: Histiocitosis; Celulas Dendríticas; Inmunodeficiencia Primaria.
Summary
Histiocytosis is a group of diseases, characterized by the proliferation of phagocytic-mononuclear system cells. Within this group is the histiocytosis of langerhans cells, which is characterized by clonal proliferation of dendritic cells, which can affect different organs and systems. The incidence is unknown being more frequent in childhood especially during the first and fourth year of life. The definitive diagnosis is established by biopsy of the lesion, in which pathological langerhans cells are observed (CD1-CD207 positive). Primary immunodeficiencies constitute a group of genetic defects, which generate alterations in both innate and adaptive defense mechanisms, that condition not only high susceptibility to infections by different infectious agents but also the alteration of hemostatic and surveillance mechanisms that prevent the development of autoinflammatory and neoplastic diseases.
Key words: Histiocytosis; Dendritic cells; Primary immunodeficiency
INTRODUCCIÓN
Entendemos por Histiocitosis a un grupo heterogéneo de enfermedades de causa desconocida, que se caracterizan por la proliferación de células del sistema fagocítico-mononuclear, (macrófagos, monocitos, células dendríticas) en diferentes órganos y sistemas (3), dicha proliferación puede ser localizada (afectando un solo órgano) o diseminada afectando varios órganos y sistemas (1). Son enfermedades poco frecuentes de predominio en la edad infantil con una incidencia anual estimada en 2 a 10 casos por millón de niños menores de 15 años, no presenta predilección por género y el pico de incidencia se encuentra entre 1 a 3 años de edad (8). Desde el punto de vista de la etiopatogenia no está claro si la Histiocitosis se presenta en el contexto de una transformación maligna, sin embargo la histopatología de las lesiones indican un proceso de desregulación inmune que sugieren la participación de citoquinas pro inflamatorias como la Interleuquina 1IL, las cuales promueven la diferenciación y migración de las células de langerhans, mientras que el factor de necrosis tumoral alfa y el factor estimulante de colonias de granulocitos promueven la diferenciación de la células hematopoyéticas en células dendríticas, en las lesiones de órganos afectados (1).
En cuanto al origen neoplásico y clonal en las células de langerhans, está sustentado en el hallazgo reciente de la presencia de la mutación BRAFV600E en el 60% de las biopsias estudiadas (8). En este sentido el diagnostico de Histiocitosis de Celulas de Langerhans, se establece mediante la observación conmicroscopia electrónica de los gránulos de birbeck, cuando la inmunohistoquimica resulta contradictoria, así como también la expresión de CD1, Proteína S100, en las células histiocitarias, confirman el diagnostico (7).
CASO CLÍNICO
Se trata de preescolar masculino de 3 años de edad, con antecedente de hospitalización por Neumonía Bilateral, cuya madre refiere enfermedad actual, cuando presenta fiebre cuantificada en 39 C, de difícil manejo, al que se asocia Tos seca ruborizante, palidez cutánea-mucosa acentuada e hipoactividad, motivo por el cual se ingresa a cargo del Servicio de Pediatría. Durante su hospitalización evoluciona tórpidamente con persistencia de la fiebre durante 48 días, contínuos, de 4 horas de duración sin patrón horario, distención abdominal, evacuaciones melenicas, adenopatías cervicales bilaterales, hepatomegalia y esplenomegalia. Al ingreso se realizaron los siguientes estudios de laboratorio:
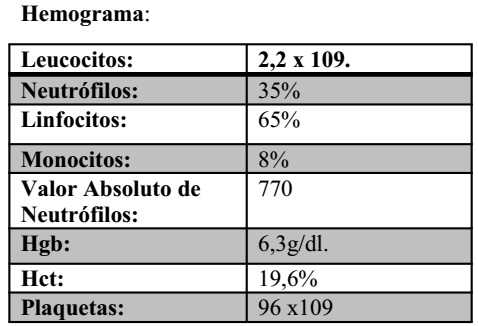
Por lo cual se determina: Leucopenia, neutropenia severa, anemia severa microcítica hipocrómica, trombocitopenia moderada.
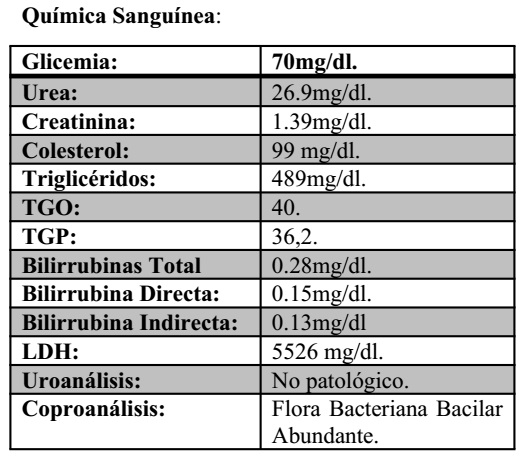
Frotis de Sangre Periférica: (Whring/ Giemsa).
- Serie Eritroide: hipocromía anisocitosis: microcitos/macrocitos, poiquilocitosis: ovalocitos, dianocitos, policromasia y eritroblastos ortocromatofilicos circulantes.
- Serie Mieloide: Polimorfonucleares con granulaciones toxicas moderadas. Linfocitos de aspecto maduro. Monocitos con vacuolas citoplasmáticas.
- Serie Megacariocitica: Anisoplaquetosis: microplaquetas y algunas macroplaquetas. Azul Brillante de Cresil: Punteado Basófilo en Eritrocitos.
Durante la hospitalización se toman muestras para estudios serológicos, hemocultivos lo cual resultan:
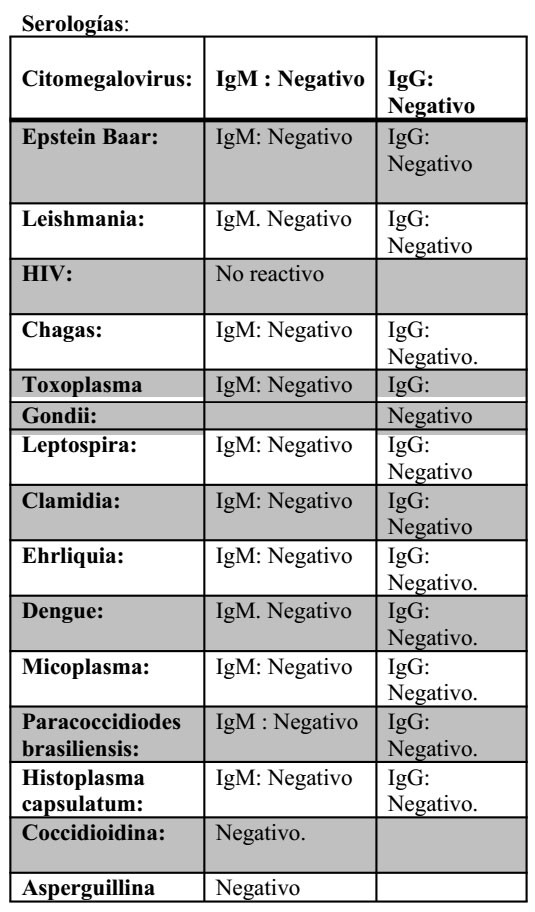
Baciloscopia de contenido Gástrico: Negativo
PPD: Negativo

- Hemocultivo: Sin crecimiento bacteriano a los 7 días de incubación.
Adicionalmente se realizan estudios imagenológicos los cuales permiten evidenciar: RX de Tórax AP: Ensanchamiento de arcos costales, broncograma aéreo, e imagen de consolidación en hemitórax derecho, adenopatías hiliares derecho, ángulos costofrenicos y costodiafragmaticos libres.
Ecosonograma Abdominal: Hígado: aspecto y configuración aumentada con vasos intrahepaticos con trayecto normal. Bazo: aspecto y configuración aumentado de tamaño 120 mm de ecogenicidad adecuada sin imágenes patológicas en su interior. Ante los hallazgos clínicos y hematológicos de citopenias periféricas se plantea la realización de aspirado y biopsia de medula ósea y se toma muestra para extendidos y se incluyen muestras para evaluación cito-morfológica, mielocultivosinmunofenotipo por citometría de flujo, y se obtienen los siguientes hallazgos morfológicosinmunológicos:
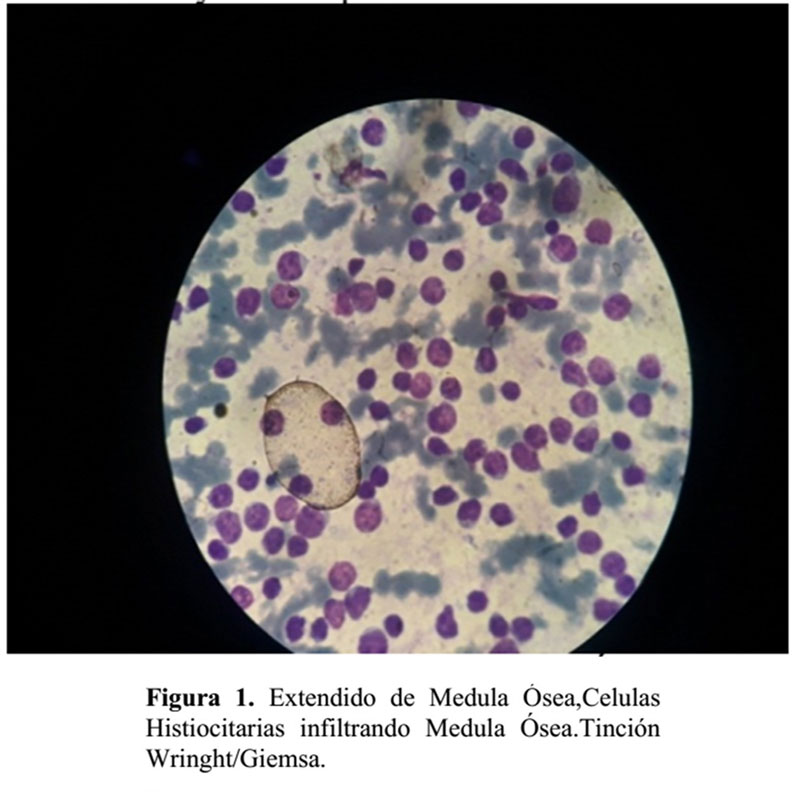
- Extendido de Medula Ósea: tinción con whring/ giemsa y evaluación mediante microscopia óptica con objetivo de inmersión se evidencia: Medula Ósea con Panhiperplasia en serie eritroide, mieloide, megacariocítica y presencia de algunos histiocitos y células plasmáticas.
- Biopsia de Medula Ósea: Hipercelular cuyo tejido hematopoyético está compuesto por 80% de promielocitos hipergranulares y 10% de linfocitos inmaduros, 10% de células mieloides inmaduras cuyos hallazgos morfológicos son sugestivos de Leucemia Promielocitica Aguda. El material de biopsia se somete a una segunda evaluación donde se obtiene: Medula ósea Hipercelular con infiltración por células histiocitarias sin atipias.
- El Inmunofenotipo de Medula Ósea por Citometria de flujo indica: Positividad para CD45+HLADR-CD34 CD13+ CD33+ CD15+ CD16+ correspondiente a una medula ósea con serie mieloide hiperplasia y serie monocitica con alteraciones importantes en la maduración antigénica. Sin criterios para Leucemia Aguda.
- Inmunohistoquimica de Medula Ósea: CD1: Inmunoreaccion positiva en células histiociticas.
Proteína S100: Inmunoreaccion positiva en células histiociticas.
CD68: Inmunoreaccion positiva en histiocitos.
Estos hallazgos inmunohistoquimica se corresponden con infiltración en medula ósea por células histiocitarias compatibles con Histiocitosis de Células de Langerhans.
Adicionalmente previo al inicio del tratamiento se evalúan las subpoblaciones linfocitarias, inmunoglobulinas y fagocitosis, los cuales se obtiene los siguientes resultados: Fagocitosis de Candida (Nitroazultetrazoil) NBT: O.37 Valor Referencia: Igual/mayor 1.70 ffl.
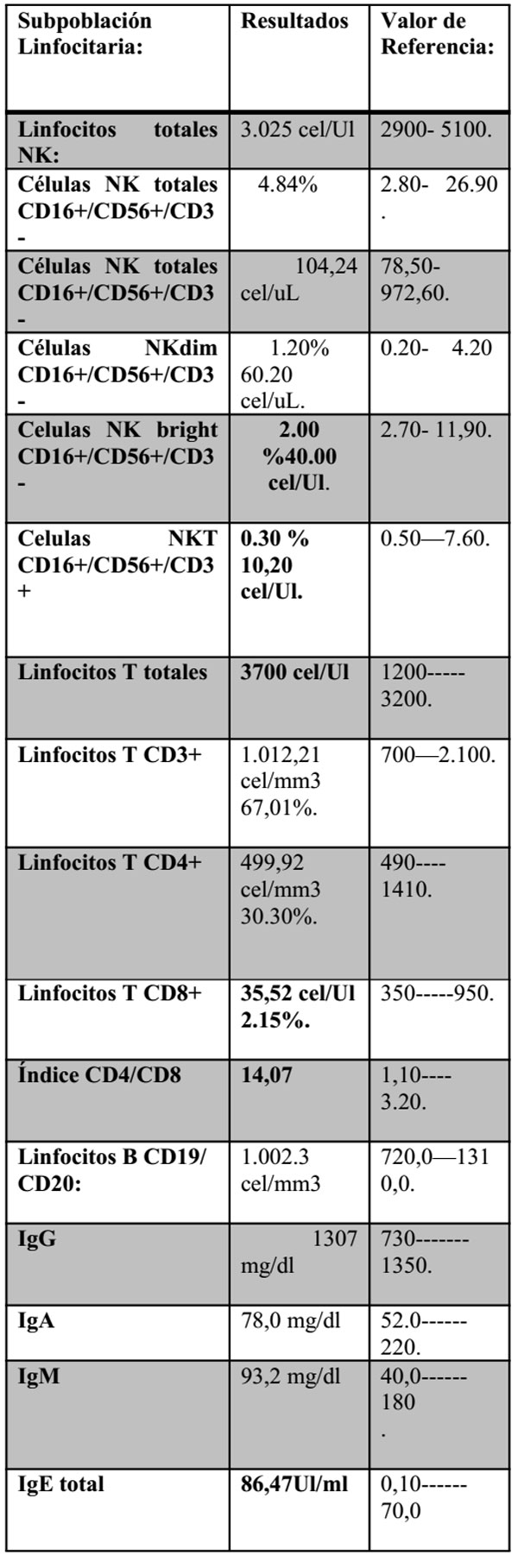
Con lo cual se establecen los siguientes Diagnósticos:
Histiocitosis de Celulas de Langerhans tipo IIA Alto Riesgo, (Infiltración Medula Ósea) asociado a Inmunodeficiencia Primaria: de Celulas T combinadas.
Se indica tratamiento con Prednisona a dosis de 1mg/kg/dosis, durante 3 meses obteniendo mejoría clínica , e inicia protocolo Histiocyte Society Langerhans Cell Histiocytosis 2009, donde cumple fase de Inducción con Vinblastina 6mg/m2 semanal durante 5 semanas + Prednisona : 40mg/m2 V.O diario durante 4 semanas con ajuste de la dosis a partir d la 4 semana + antibioticoterapia profiláctica, obteniendo remisión hematológica y manteniéndose clínicamente asintomático, seguidamente cumple fase de mantenimiento la cual inicia desde la semana 7 de post inducción, con pulsos de Vinblastina, Prednisona cada 21 días, Mercaptopurina continuo hasta la semana 14 cuando reaparece fiebre persistente durante 28 días, hepatomegalia, esplenomegalia, pancitopena: anemia moderada microcitica – hipocromica, leucopenia, neutropenia severa, trombocitopenia leve, se ingresa como una reactivación /progresión de la histiocitosis.
Durante la hospitalización tiene evolución tórpida, asociándose disnea, tos seca, distención abdominal, oliguria, disfunción hepática, diátesis hemorrágicas, falla multiorgánica, lo cual sugiere complicación con probable síndrome de activación
macrofágica.
DISCUSIÓN
La Histiocitosis se caracteriza por una activación patológica del sistema inmune. La forma de presentación del caso se explica en el contexto de un origen primario que, aunque un evento infeccioso puede asociarse como un desencadenante, existe otro evento de tipo genético desconocido que incluye una inmunodeficiencia primaria la cual actuaría como el factor determinante que impulsa junto a factores exógenos la aparición y las exacerbaciones de la enfermedad.
La positividad en las células histiocitarias para CD1, s-100, CD68, confirman el origen de las células como de linajedendrítico, (Celulas de Langerhans), que infiltran medula ósea, tejido hematopoyético, estableciéndose como de Alto riesgo. La deficiencia de células NK, linfocitos CD8+ fagocitosis, constituyen la base de una inmunodeficiencia primaria, combinada de células T.
En este sentido los aspectos patogénicos evaluados reflejan que una deficiencia en las funciones de los linfocitos T, en la citotoxicidad mediada por los linfocitos CD8+ inducen la activación exagerada y persistente de células presentadoras de antígenos (células dendríticas, macrófagos), linfocitos CD4+, CD8+ con la consecuente liberación de citoquinas pro inflamatorias entre ellas el FNTa, Interleuquina IL1, la cual promueven la diferenciación , proliferación y migración de células de langerhans, a los diferentes órganos (hueso , piel, pulmón, bazo, hígado, medula ósea, ganglios linfáticos). El establecimiento agudo del síndrome de activación macrofagica como una complicación brusca durante el tratamiento, caracterizada por fiebre persistente, hepatoesplenomegalia, citopenias, disfunción hepática y renal, afectación neurológica, coagulación intravascular diseminada, se desarrollan en el contexto de la tormenta de citoquinas, impulsadas por falla importante en la regulación inmune, activación de linfocitos, fracaso en las funciones citotóxicas de linfocitos CD8+, hipersecreción de citoquinas y activación macrofágica con la consecuente hemofagocitosis en órganos como bazo, hígado, medula ósea, pulmón y el establecimiento de falla multiorgánica.
Referencias
1. Haup t R, Minkov M, Astigarraga I et al., Langerhans cell histiocytosis (lch): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years, Pediatr Blood
Cancer 2013; 60(2):175-84.
2. Emile JF, Abla O, Fraitag S et al., Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages, Blood 2016; 127(22):2672-81.
3. Zinn DJ, Chakraborty R y Allen CE, Langerhans cell histiocytosis: emerging insights and clinical implications, Oncology (Williston Park)2016; 30(2):122-32, 139.
4. Badalian-Very G, Vergilio JA, Degar BA et al., Recurrent braf mutations in Langerhans cell histiocytosis, Blood 2010; 116(11):1919-23.
5. Coury F, Annels N, Rivollier A et al., Langerhans cell histiocytosis reveals a new il-17a-dependent pathway of dendritic cell fusion, Nat Med 2008; 14(1):81-7.
6. Willman CL, Busque L, Griffith BB et al., Langerhans’-cell histiocytosis (histiocytosis x): a clonal proliferative disease, N Engl J Med 1994; 331(3):154-60.
7. Zeng K, Ohshima K, Liu Y et al., braf v600e and map2k1 mutations in Langerhans cell histiocytosis occur predominantly in children,
Hematol Oncol2016; Epubenpreparación.
8. Nelson DS, Quispel W, Badalian-Very G et al., Somatic activating araf mutations in Langerhans cell histiocytosis, Blood 2014; 123(20):3152-5.
9. Gadner H, Heitger A, Grois N et al. Treatment strategy for disseminated Langerhans cell histiocytosis. DAL HX-83 Study Group. Med PediatrOncol. 1994; 23: 72-80.
10. Gadner H, Grois N, Potschger U et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy
intensification. Blood. 2008; 111: 2556-2562.
11. Weitzman S, Braier J, Donadieu J et al. 2′-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH).
results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer. 2009; 53: 1271-1276.
12. Minkov M, Steiner M, Potschger U, et al. Reactivations in multisystem Langerhans cell histiocytosis: data of the international LCH registry. J Pediatr. 2008;153:700-5,705.e1-2.
13. Pollono D, Rey G, Latella A, et al. Reactivation and risk of sequelae in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007;48:696-
699.

Autores
Dr. José de J. Ledezma F
Adjunto de Unidad de Hematología. Hospital General
Dr. Israel Ranuarez Balza. San Juan de los Morros, Estado Guárico.
- Este autor no ha escrito más artículos.