
Aplasia medular severa asociado a desorden linfoproliferativo no clonal de celulas T grandes granulares. reporte de un caso
RESÚMEN
La Anemia aplásica, es una enfermedad hematológica no maligna, poco frecuente que forma parte de los síndromes de falla medular, caracterizada por pancitopenia periférica y medula ósea hipocelular. La etiología puede ser de forma adquirida, primario o idiopático lo cual representa el 70 %-80% de los casos o secundario a infecciones, medicamentos, tóxicos, irradiación. Es una enfermedad heterogénea donde la susceptibilidad genética y la destrucción inmunomediada de la célula progenitoras hematopoyéticas juegan un papel primordial. El síndrome linfoproliferativo de células T granulares, forman parte de un grupo heterogéneo de desórdenes linfoproliferativos no clonales caracterizado por la persistencia en sangre de linfocitos de aspecto granular, asociada a citopenias. Tiene un curso indolente no progresivo y puede desarrollarse en el contexto de situaciones infecciosas, autoinmunes, neoplásicas. Desde el punto de vista clínico se presenta en el contexto de una aplasia medular, o asociado a hepatoesplenomegalia, y citopenias en sangre periférica.
Palabras Clave: anemia aplásica, leucemia de células t grandes granulares, linfocitosis.
SUMMARY
Aplasic anemia is a non-malignant hematological disease, which is rare in spinal cord syndromes, characterized by peripheral pancytopenia and hypocellular bone marrow. The etiology can be acquired, primary or idiopathic, which represents 70% -80% of cases or secondary to infections, medications, toxic, irradiation. It is a heterogeneous disease where genetic susceptibility and immune-mediated destruction of hematopoietic progenitor cells play a key role. Granular T-cell lymphoproliferative syndrome is part of a heterogeneous group of non-clonal lymphoproliferative disorders characterized by the persistence in blood of lymphocytes of granular appearance, associated with cytopenias. It has an indolent non-progressive course and can develop in the context of infectious, autoimmune, neoplastic situations. From the clinical point of view it is presented in the context of spinal cord aplasia, or associated with hepatosplenomegaly, and cytopenias in peripheral blood.
Key words: Aplasic anemia, granular large t-cell leukemia, lymphocytosis
INTRODUCCIÓN
La anemia aplásica, es una enfermedad hematológica no maligna, poco frecuente, que forma parte de los síndromes de falla medular, caracterizada por pancitopenias periféricas y medula ósea hipocelular de forma persistente, en ausencia de signos morfológicos displásicos y fibrosis.(2) Desde el punto de vista etiológico, puede tratarse de un problema adquirido primario o idiopático, lo cual representa el 70%- 80% de los casos, secundario a infecciones, medicamentos, tóxicos, irradiación o congénita (2). La incidencia de aplasia medular en niños se estima de 2 casos por cada millón de habitantes en países orientales. En Venezuela, no contamos con un registro confiable de los casos (5).
La patogénesis de esta enfermedad es muy heterogénea y es considerada como una enfermedad inmunomediada en la cual la desregulación de linfocitos citotóxicos CD8+.CD4+NK, junto a la producción anormal de citoquinas tales como interferón gamma (IFN-Y), factor de necrosis tumoral alfa (TNF-a), orquestan la destrucción y apoptosis de la célula madre hematopoyética; en definitiva, una gran cantidad de datos clínicos y de laboratorios sugieren que la supresión inmunomediada a través de la expansión oligoclonal de linfocitos T autoreactivos inducen la apoptosis del stem cell, estableciéndose verdaderos desordenes linfoproliferativos clonales o no clonales como la proliferación no leucémica de células t grandes granulares ( T LGL ) reactiva, y fase leucémica (LGL), lo cual constituyen un grupo heterogéneo de desórdenes caracterizado por linfocitosis persistente asociado a citopenias periféricas, cuyo linfocito t , presenta morfología granular, fenotipo CD8+, CD4+, NK respectivamente (2).
Es un desorden de curso indolente, no progresivo, y se presenta en el contexto varias enfermedades como infecciosas, autoinmunes, falla medular, entre otros (4). Desde el punto de vista de la patogénesis la (T LGL), no leucémica puede derivarse de proliferaciones no clonales oligo y policlonales de subpoblaciones de linfocitos T con cambios evolutivos en la clona dominante, mientras que en la forma leucémica (LGL), las células se tornan resistentes a la apoptosis y expresión aberrante STAT3 activado (6). En este contexto se describe el siguiente caso de preescolar con de aplasia medular asociado a desorden linfoproliferativo no clonal de células T grandes granuales, como una forma de expresión inusual desde el punto de vista clínico, morfológico e inmunológico en las anemias aplásicas.
DESCRIPCIÓN DEL CASO
Se presenta caso de preescolar masculino de 4 años de edad, sin antecedentes patológicos de base conocidos, procedente de Guárico, quien inicia enfermedad actual hace 7 meses cuando comienza a presentar palidez cutáneo mucosa, debilidad generalizada por lo que acude a facultativo e indican paraclínicos que reportan hemoglobina en 4,8 gr/dl, hematocrito 15% motivo por el cual se ingresa.
Paciente quien al ingreso se encuentre en regulares condiciones generales con palidez cutánea mucosa acentuada y ligero tinte ictérico.
Se realiza hemograma que reporta lo siguiente:
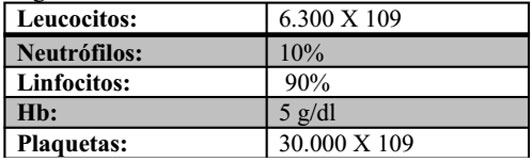
Resultados: Linfocitosis absoluta, neutropenia severa, anemia severa, trombocitopenia moderada.
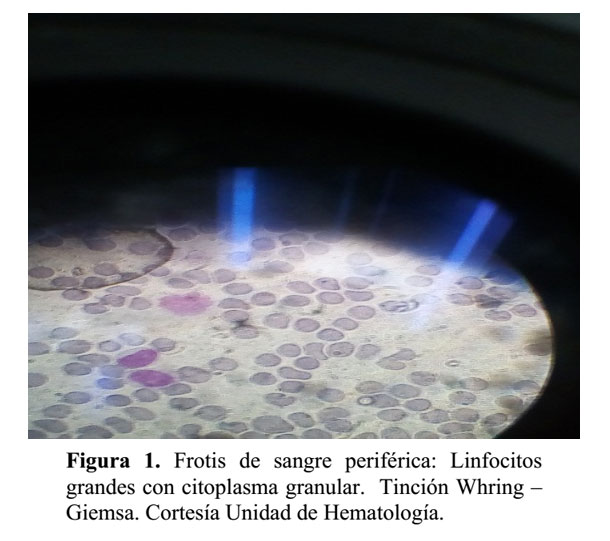
Frotis de Sangre Periférica:
Serie eritroide: Hipocromía: ++ , anisocitosis: +.
Serie Mieloide: Neutrófilos disminuidos.
Linfocitos: Aumentados, constituidos por una población heterogénea, conformada por linfocitos de aspecto maduro, linfocitos grandes que presentan núcleo regular, cromatina madura, citoplasma ligeramente basofílico que contienen en su interior granulaciones finas, azurofíilicas dispuestas hacia la periferia.
Serie Megacariocítica: Plaquetas disminuidas, constituidas por microplaquetas que conforman pequeños agregados, de morfología hipogranular (plaquetas grises).
Se realiza Aspirado y biopsia de cedula ósea, se obtiene muestra para extendidos, evaluación cito-morfológica para evaluación histopatológica obteniendo los siguientes hallazgos:
1.- Extendido de Medula Ósea: Hipocelular, celularidad escasa, disminuida para la edad: 30%.
Serie eritroide: disminuida con maduración normoblastica.
Serie Mieloide: disminuida con arresto parcial en la maduración. Serie Megacariocítica: Megacariocitos disminuidos, hipolobulados, pocos productivos.
Se observa infiltración por linfocitos de aspecto maduro, que se disponen en agregados aislados. Tejido adiposo presente, aumentada en relación con el tejido hematopoyético. No se observó infiltración por leucemia ni linfoma.
Los hallazgos morfológicos son sugestivos de probable aplasia medular a correlacionar con clínica y biopsia del paciente.
2.- Biopsia de Medula Ósea: Contenido hematopoyético marcadamente reducido en relación a otros elementos de la medula, serie eritroide, mieloide, megacariocítica notablemente disminuida limitada a pequeños grupos celularesentre tejido adiposo, sugestivos de Aplasia Medular.
3. Inmunofenotipo de Medula Ósea: Indica positividad para CD45 + CDD3+ CD8+ CD20+CD34-.
Los hallazgos inmunofenotípicos de muestra de medula ósea indican: Medula ósea sin infiltración patológica, serie mieloide disminuida, con incremento de la población linfoide, no se evidencian células inmaduras ni precursores hematopoyéticos, dichos hallazgos son compatibles con insuficiencia medular (Síndrome de falla medular).
En tal sentido los aspectos clínicos, hallazgos morfológicos, histopatológicos e inmunofenotípicos son concluyentes para: Desorden proliferativo no clonal de linfocitos T grandes granulares (L-LGL) complicado con aplasia medular severa
En este contexto se inicia tratamiento de primera línea con:
- Ácido Fólico 5mg O.D,
- Prednisona: 1mg/kg/día,
- Revolade 25mg OD durante dos meses; realizando solicitud para tramites de trasplante alogénico de medula ósea.
Paciente quien no obtiene remisión hematológica, evolucionando de manera tórpida, asociándose neutropenia severa, anemia severa y trombocitopenia severa con manifestaciones hemorrágicas, recurrentes mucocutáneas, hematurias macroscópicas, ameritando múltiples hospitalizaciones para su manejo, apoyo transfusional continuo, por lo cual, en vista de complicaciones y progresión de la enfermedad se decide previa evaluación serológica para toxoplasmosis, HIV, hepatitis B, citomegalovirus, epstein barr y hongos; instaurar tratamiento con protocolo (TIS) inmunosupresor:
- Ciclosporina 15mg/kg/dosis continuo
- Metilprenisolona (pulsos) 1mg/kg/día, durante 4 días
- Prednisona: 1mg/kg/día durante 17 días
- Factor estimulante de colonias granulocitos
- Eritroproyetina: semanal
- Apoyo transfusional plaquetario profiláctico sos plaquetas <20 x 109
- Antibioticoterapia profiláctico: trimetroprin/ sulfametoxazol, fluconazol y aciclovir.
8) Infusión de ATGAM (Globulina Antitimocito de Caballo) a dosis de 40mg/kg durante 3 días, continuación de Ciclosporina 15mg/kg/día, Metilprenisolona 1mg/kg/día durante 3 días lo cual culmina el 2/12/19.
Obteniendo respuesta parcial el día 9 de reposo de infusión caracterizado por aumento de cifras de neutrófilos y plaquetas, disminución del valor absoluto de linfocitos y 14 días sin requerimiento transfusional.
DISCUSIÓN
El síndrome de falla medular, se define como una producción disminuida de uno o más linajes hematopoyéticos, la forma de presentación es congénita o la anemia aplasica adquirida, la cual se caracteriza por pancitopenia periférica y medula ósea hipocelular con diferentes grados de severidad.
En cuanto al agente etiológico del caso, adjudica a la de causa idiopática, donde no se identifican factores de riesgo, como lo es exposiciones a agentes tóxicos, medicamentos, radiaciones e infecciones 6 meses previos a la aparición de la enfermedad. Sin embargo, en los mecanismos que disminuyen la hematopoyesis y aparece la pancitopenia periférica en la cual se pone en evidencia la destrucción inmunomediada del stem cell, están bien dilucidados en el caso representados por los siguientes criterios morfológicos, histológicos e inmunofenotipicos:
- Linfocitos cuya población presenta morfología heterogénea, núcleo de cromatina madura, algunos con citoplasma moderado y granulaciones finas azurofílicas en su interior. El Inmunofenotipo de muestra de medula ósea indica positividad para CD45 + CDD3+ CD8+ CD20+ concluyente para infiltración ósea por linfocitos fenotipo TCD3+CD8+CD4- de aspecto maduro.
- Linfocitosis absoluta (98%) persistente >6 meses.
- Citopenias periféricas: neutropenia severa (VAN:400 cel.), anemia severa, trombocitopenia severa persistente >6 meses.
- Linfocitos maduros de morfología granular.
- Infiltrado de linfocitos en medula ósea y sangre periférica fenotipo de células T CD3+CD8+CD4+.
Lo cual indica que los hallazgos clínicos, morfológicos, fenotípicos son sugestivo de probable: Desorden proliferativo no clonal de linfocitos T grandes granulares (L-LGL) complicado con aplasia medular severa. En cuanto a los mecanismos que subyacen al establecimiento del desorden linfoproliferativo no clonal de células T granulares se presentan en el contexto de factores autogénicos, antígenos virales, que pueden llevar a la activación crónica de los linfocitos T CD8+. En este sentido las citopenias se desarrollan en el caso de la neutropenia por destrucción directa de precursores mieloides, alteración en la maduración de los mismos y destrucción periférica como consecuencia de la inducción de la apoptosis, mientras que la anemia y trombocitopenia se debe por inhibición de los precursores en medula ósea, destrucción periférica por secuestro esplénico.
REFERENCIAS
- Liu X, LougharanJr , The spectrum of large granular lymphocyte leukemia and feltys syndrome curr .OpinHematol. 2011:18:254-9
- Sokol L, Lougharan PT Jr, Large Granular Lymphocyte Leukemya. Oncologist, 2006;11(3): 263-73. 3.
- Lougharan PT Jr. Clonal Diseases of large granular lymphocytes. Blood.1993;82(1):1-14. 4.
- LamyT,LougharanTPJr.Clinical Features of Large granular lymphocytes Leukymia.Semin Hematol.2003;40(3)185-95. 5.
- Spling Burnette BK, Lougharan PT Jr. Survival signals in Leukymia large granular Limphocytes.Semin Hematol;40(3)213-20. 6.
- Marsh JCW,etal.Guidelines for the diagnosis and management of aplasicanaemia. Br Haematol. 2009;147:43-70. 7.
- Sheinberg P, Young NS, How I treat acquired aplastic anemia.Bood. 2012;120(6):1185-1196. 8.
- Guinan EC. Diagnosis and management of aplastic anemia. Hematology. 2011 76-81. 9.
- Sheinberg P, et al. Predicting response to inmunossuppresive therapy and survival in severe aplastic anaemia.Br Haematol.2008 144:206-216. 10.
- Gupta V. et al. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anaemia using HLA matched sublingdonors.Haematologica.2010;95 (12):2119-2125
