
Migraña, revisión actualizada
Ciertamente, aún queda mucho por aclarar sobre cómo entendemos la migraña. Ha afectado a los humanos durante más de dos milenios, en todos los continentes estudiados y con más de mil millones de pacientes que sufren al menos una crisis dolorosa cada año. Esta enfermedad se erige como la sexta causa más común de discapacidad en el planeta. La migraña es una afección cerebral grave e incapacitante. Desafortunadamente, su clasificación aumenta con el tiempo. Tiene una prevalencia anual entre el 15 al 18% en todo el mundo(1). Si se incluyen tanto la migraña episódica como la crónica representa una enorme carga para las economías mundiales. La migraña afecta predominantemente a las mujeres, 3:1 y afecta significativamente la calidad de vida(2), en muchos casos durante los años pico de productividad.
La migraña es un trastorno complejo, básicamente hereditario, de la función cerebral. Muchas de las ideas en torno a su fisiopatología son relevantes, desde una predisposición genética, pasando por la hiperexcitabilidad cerebral, hasta la sensibilización periférica y central y la disfunción del tronco encefálico y el hipotálamo.
Desde el punto de vista genético, los familiares inmediatos de los migrañosos tienen un riesgo sustancialmente mayor de sufrir migraña en comparación con los familiares de los controles asintomáticos(43). Curiosamente, el riesgo relativo difiere según la presencia del aura y el grado de discapacidad. Los familiares de primer grado de pacientes que padecían migraña con aura tienen un riesgo relativo 4 veces mayor de migraña, mientras que los familiares de pacientes con migraña sin aura muestran un aumento del riesgo de solo 1,9 veces(44). Los estudios de gemelos proporcionan más pistas que revelan una tasa de concordancia de migraña significativamente más alta en gemelos monocigóticos en comparación con gemelos dicigóticos.
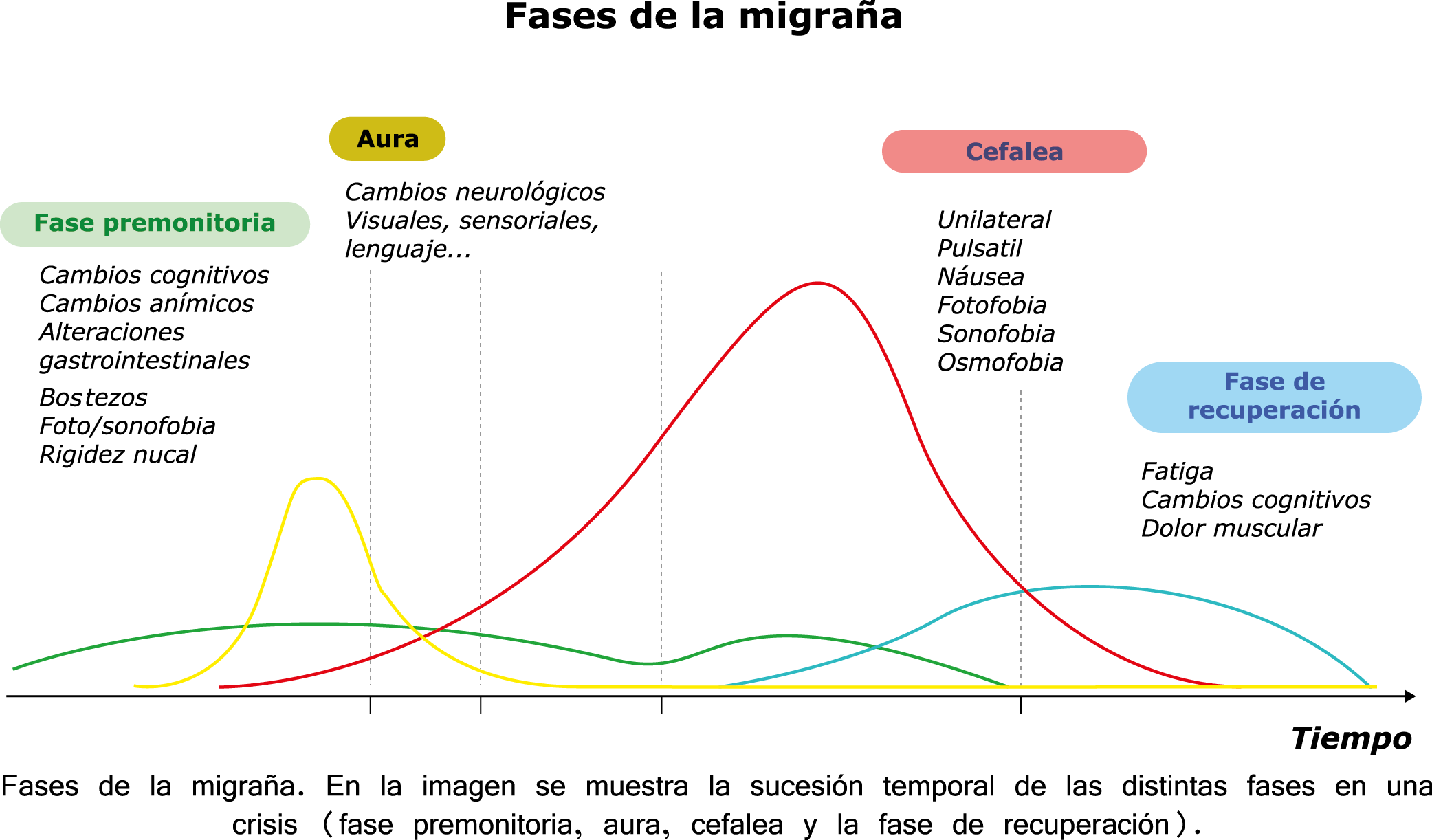
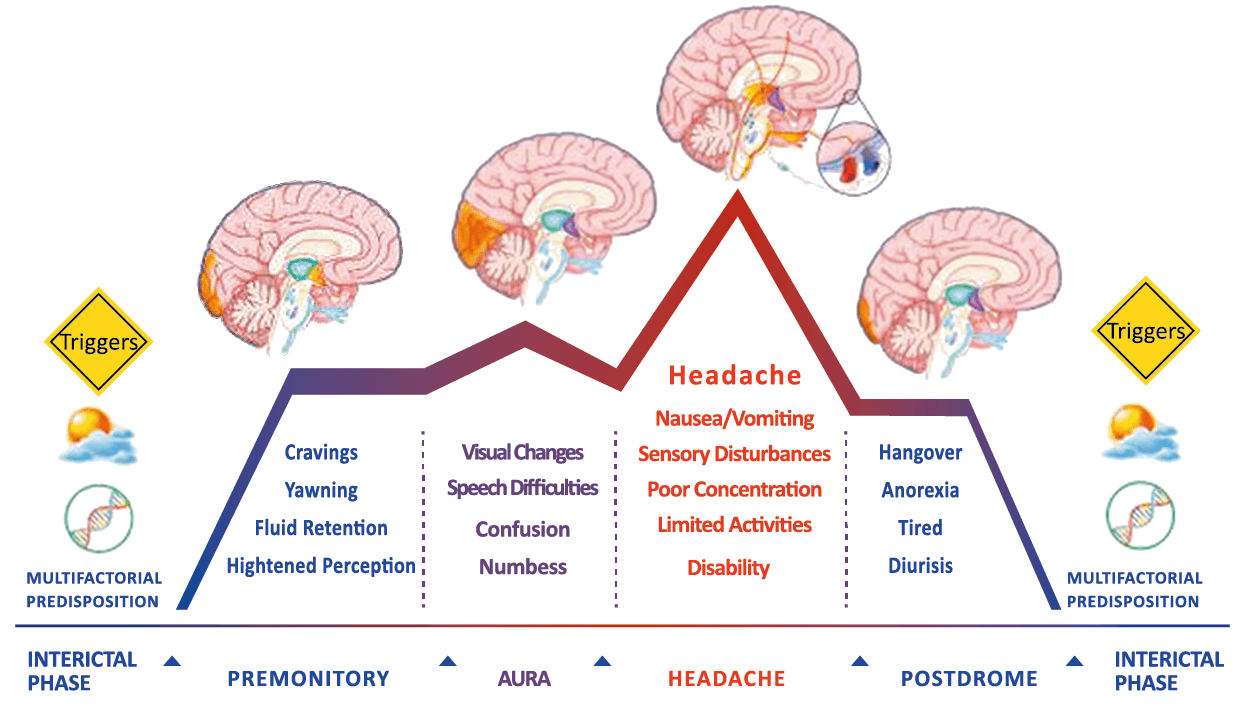
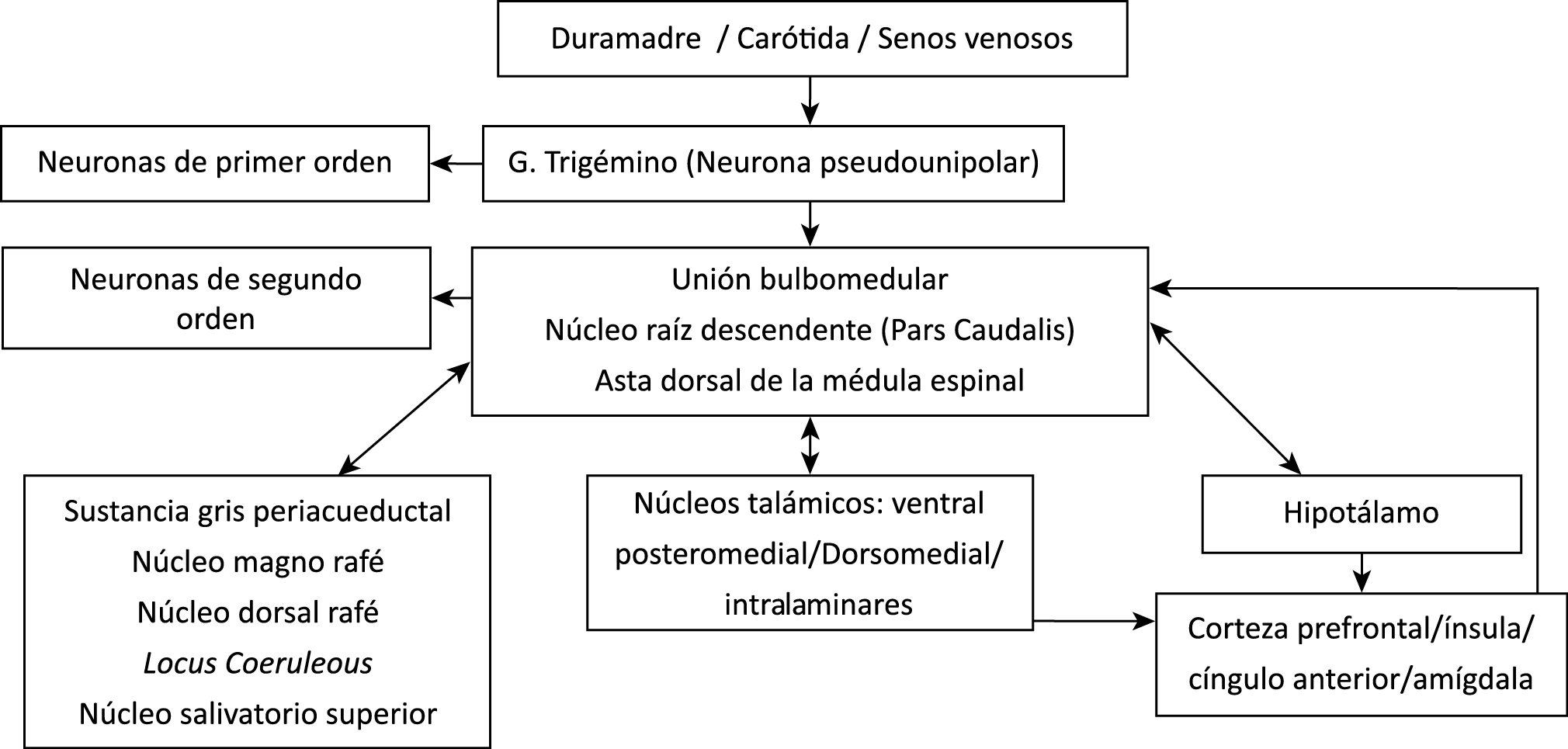
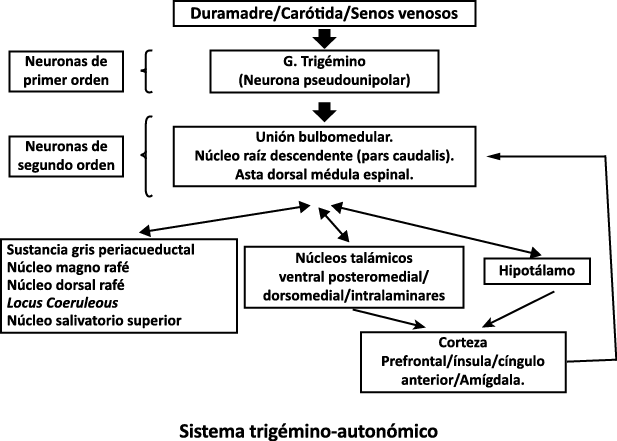
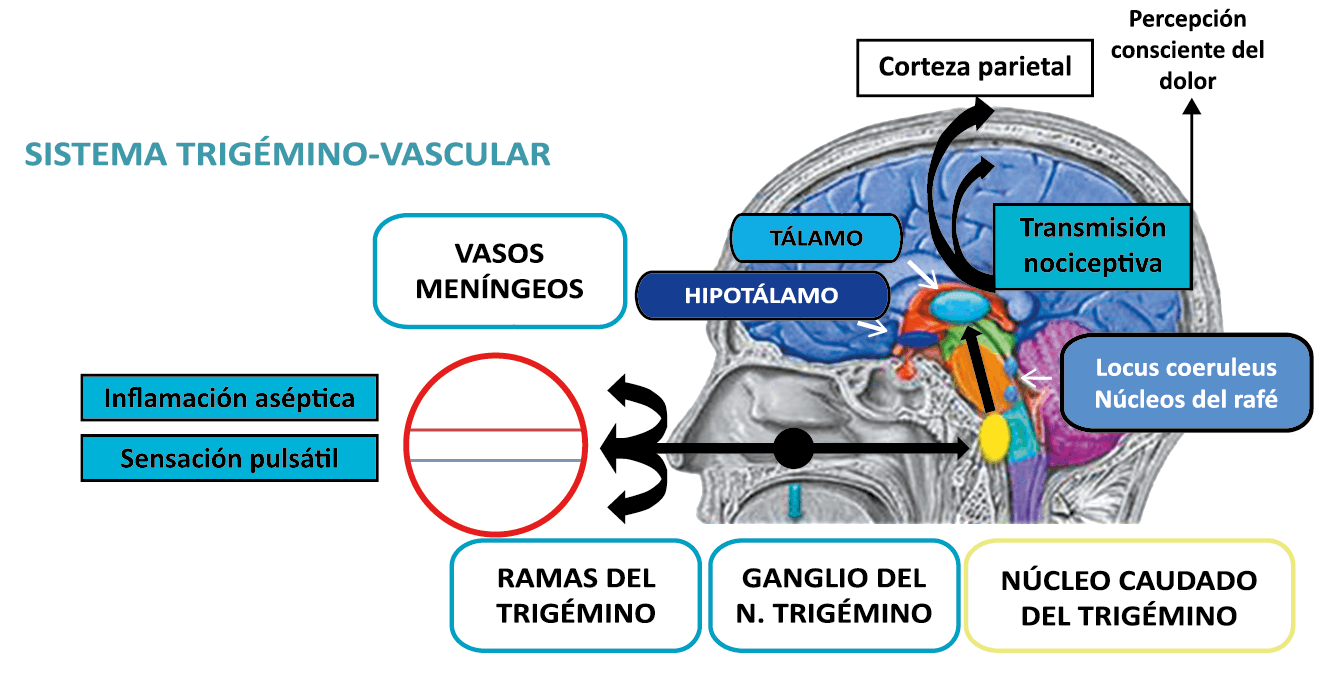
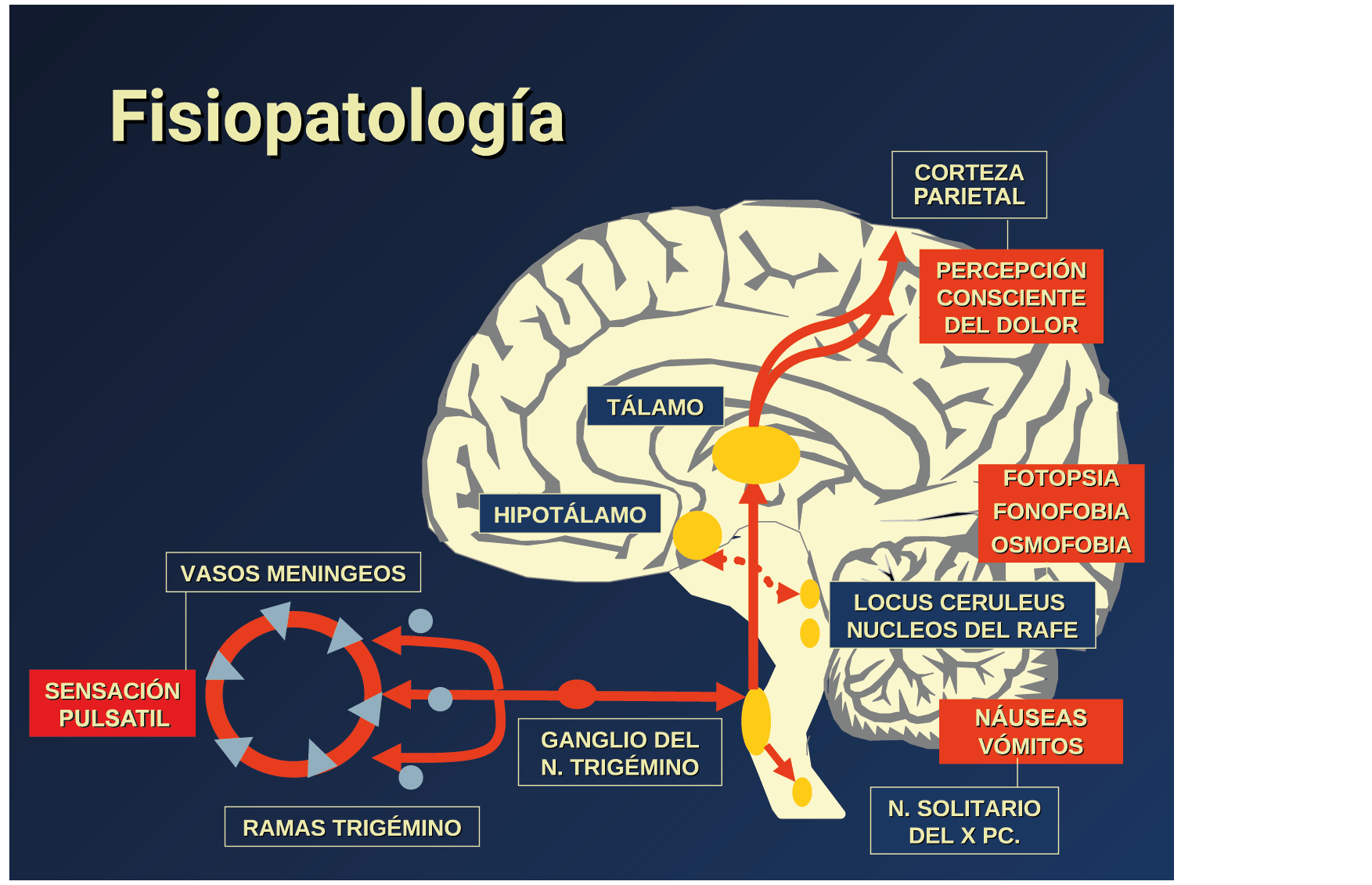
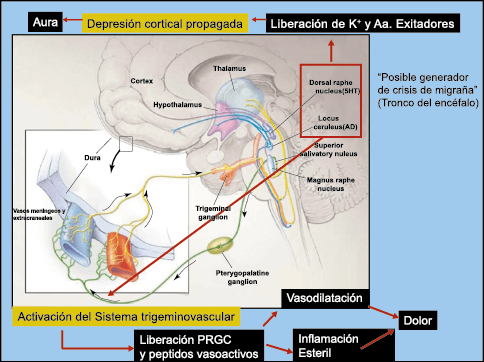
Todo lo mencionado contribuye al fenotipo del migrañoso y son vías que continuamente se están explorando para el desarrollo de nuevas terapias agudas y preventivas del dolor, más seguras y eficaces. Sin embargo, existe una controversia que gira en torno a dos cuestiones: la iniciación y el origen del dolor además de que todavía no se conoce el origen de los mecanismos neuronales que subyacen a la condición primaria en las personas susceptibles. Definitivamente la migraña implica la activación y sensibilización de las vías trigeminovasculares, así como el tronco cerebral y los núcleos diencefálicos(88). La vía aferente comienza en las aferentes vasculares nociceptivas de la duramadre que están para advertir, no para localizar. Su proyección al tálamo y la corteza y como se regula esta vía en cada nivel y por múltiples sistemas, ofrece la posibilidad de comprender los síntomas complejos y orientar las terapias. Los ataques comienzan como síntomas premonitorios, dificultad para concentrarse, bostezos, fluctuaciones del humor; y cambios homeostáticos, como la alimentación y el equilibrio de líquidos, que pueden incluir sensibilidades más generalizadas, como fotofobia y fonofobia. La fase premonitoria da paso a la fase de dolor con el sufrimiento acompañante y termina en una fase posdrómica de sentirse agotado por la experiencia. (Figura 1)
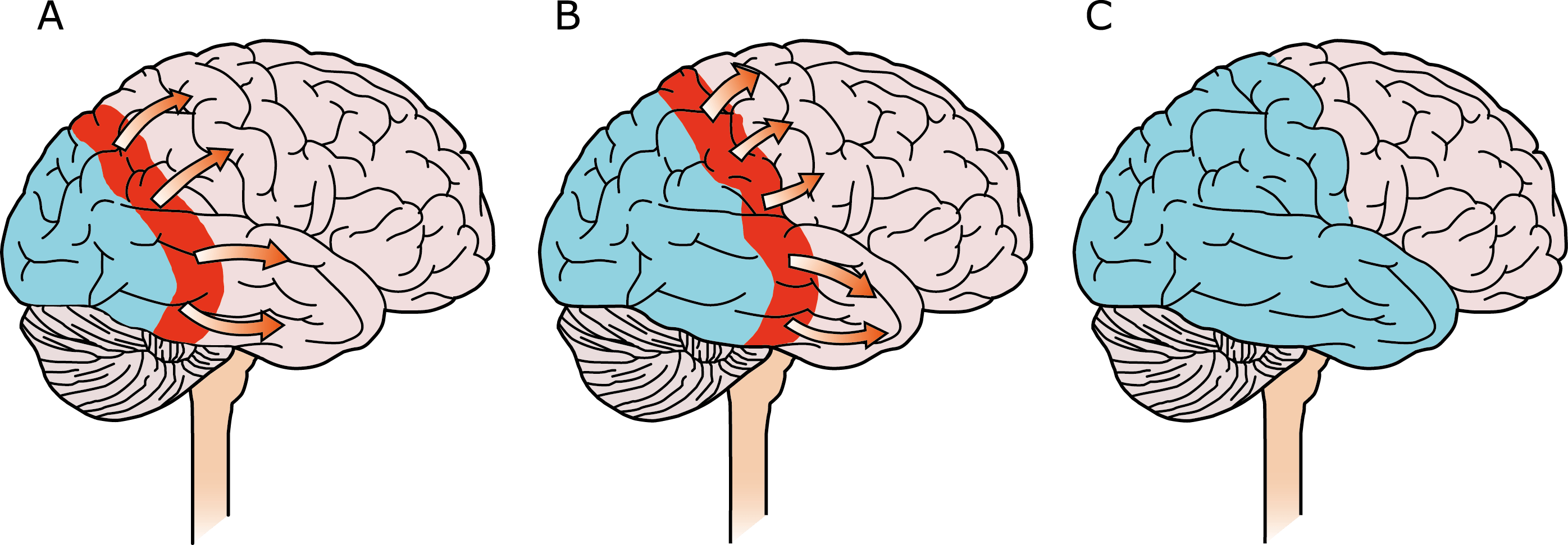
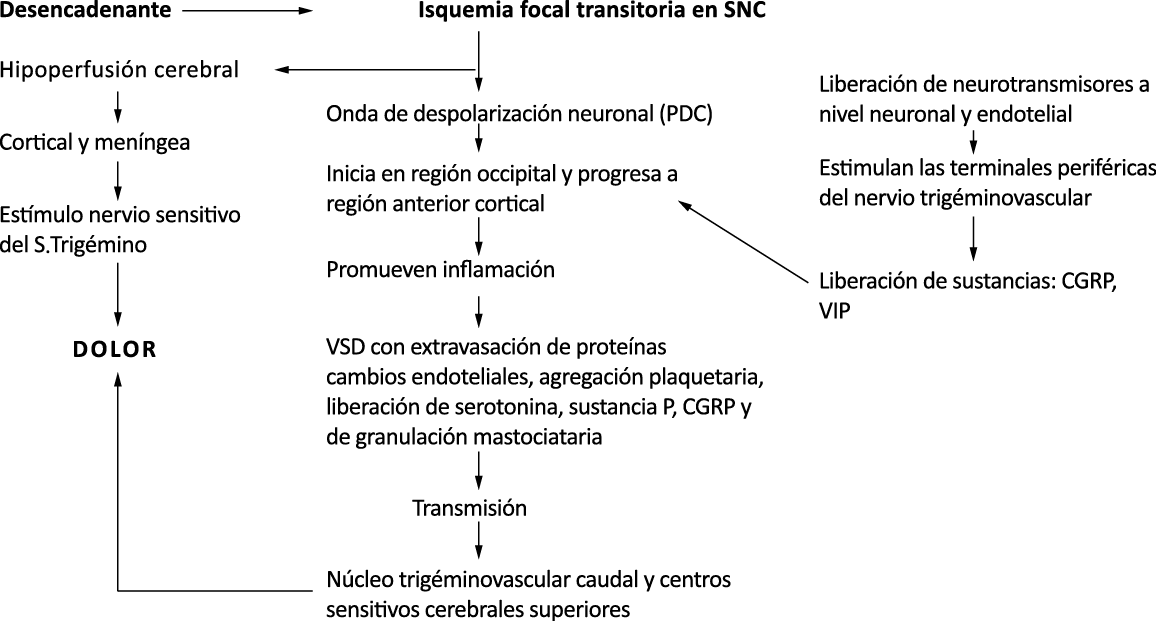
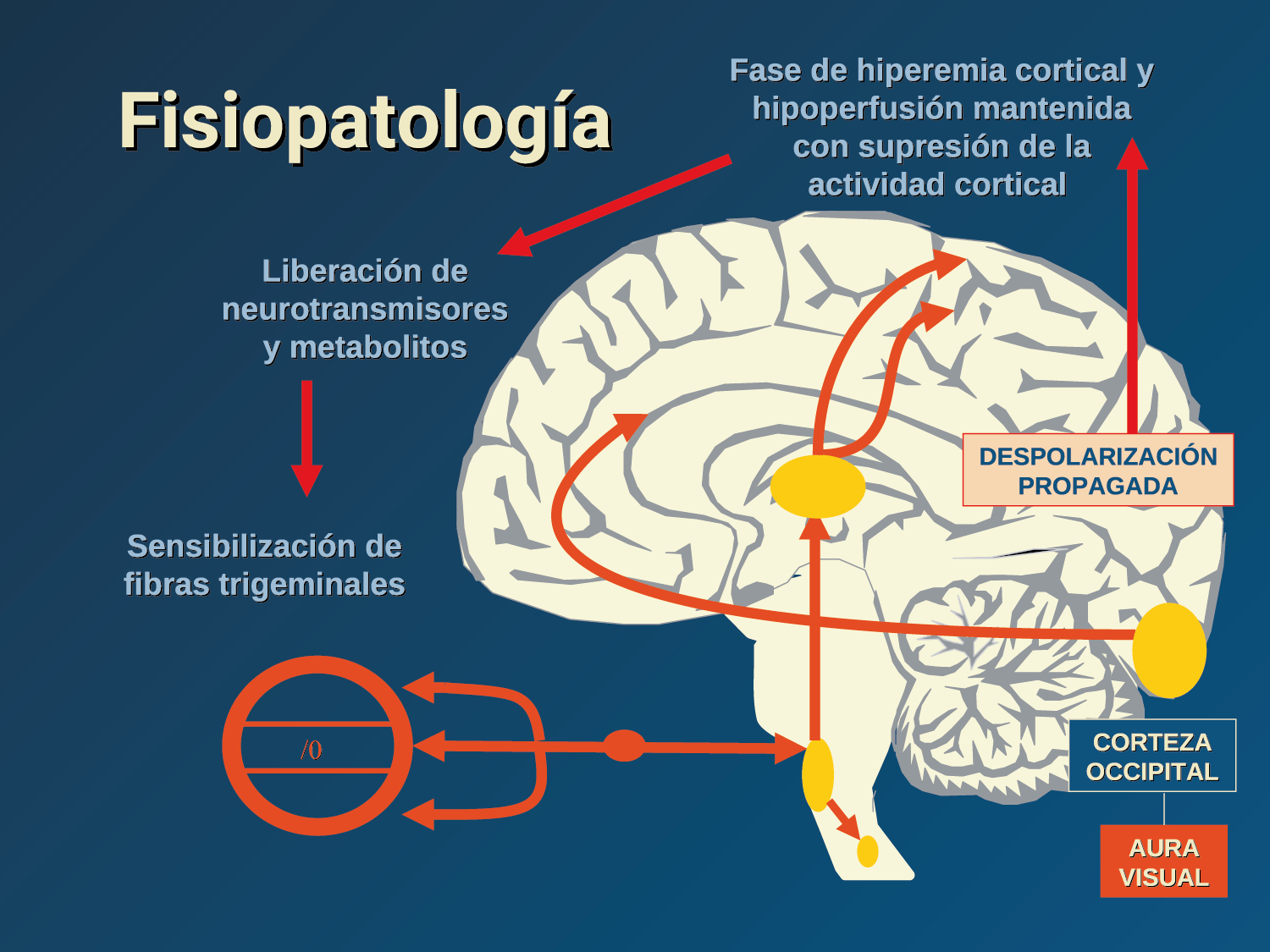
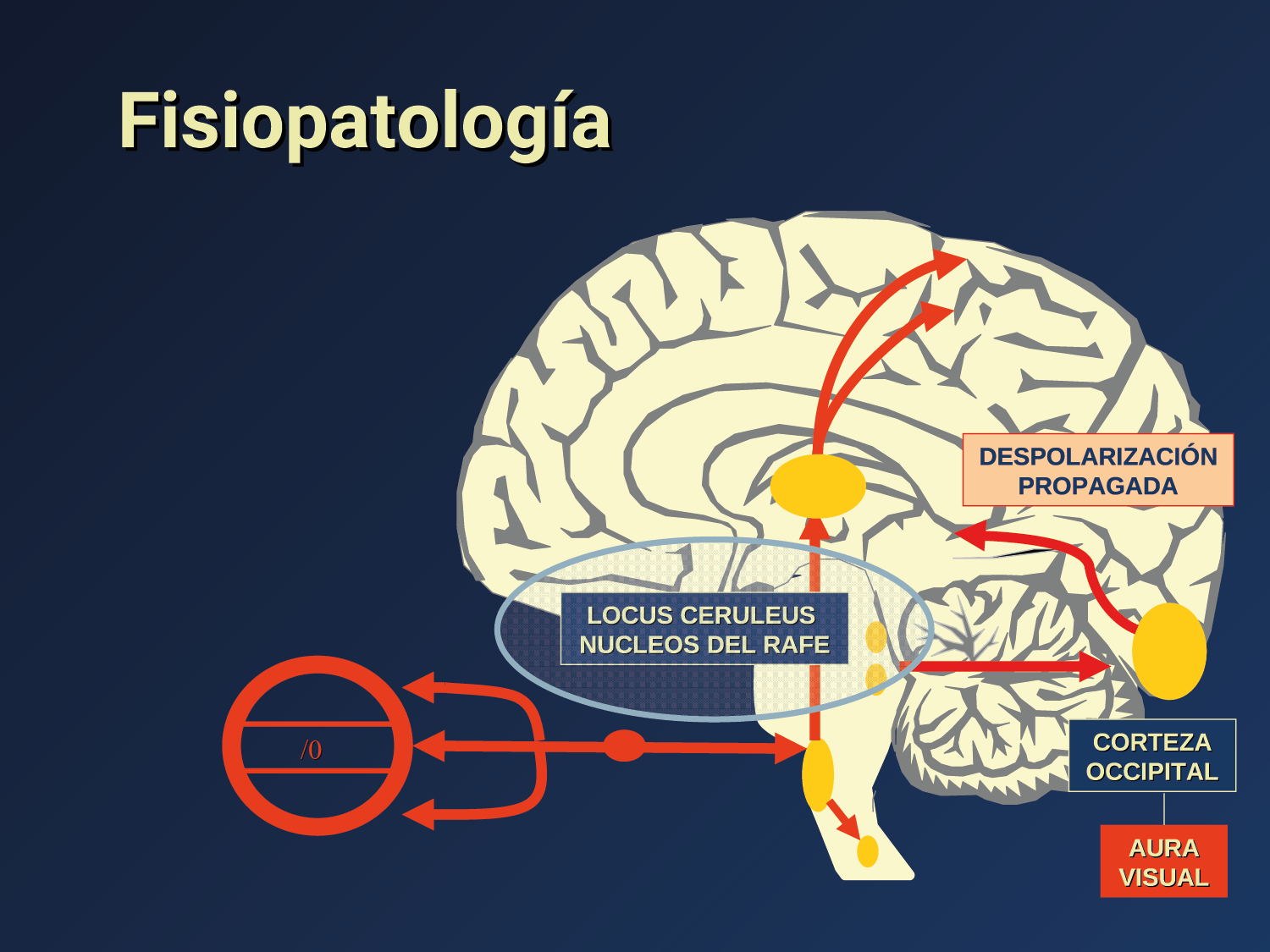
El aura de la migraña, al menos en su sentido clásico, tiene relación con la depresión cortical que se propaga por la corteza cerebral y participa como un actor paralelo cuando se producen las crisis. (Figura 2)
Un avance crucial en la comprensión de la migraña ha sido su revalorización como un trastorno predominantemente neural y el desarrollo de estrategias de tratamiento agudas y preventivas no solo vasoactivas. Comprender la migraña ofrece el beneficio tangible de reducir la carga del sexto problema médico más incapacitante del planeta. La compresión de la génesis y desarrollo de la migraña ha recorrido un largo camino en los últimos dos milenios y parece estar lista para acelerar el ritmo y brindar aún más beneficios en las próximas décadas.
La fisiopatología de la migraña había surgido de una consideración histórica de los “humores” hasta mediados del siglo XX con la ahora desaparecida Teoría Vascular. Pero aún se podría decir que hay tres preguntas: ¿por qué, cómo y cuándo? Por qué: se acepta en gran medida que la migraña es una tendencia heredada del cerebro a perder el control de sus vías de acceso de impulsos. Cómo: la ahora clásica vía aferente nociceptiva vascular de la duramadre del trigémino ofrece una hoja de ruta del ataque. Cuándo: los ataques de migraña surgen debido a un trastorno del procesamiento sensorial del cerebro que probablemente sea cíclico, influenciado por la genética y el medio ambiente.
En la primera fase (figura 3), premonitoria, que precede a la cefalea, los sistemas del tronco encefálico y diencefálico modulan las señales aferentes, la fotofobia a la luz o la fonofobia al sonido, comienzan a disfuncionalizarse y eventualmente a evolucionar a la fase de dolor y con el tiempo a la fase de resolución o posdrómica. Aproximadamente en un tercio de los pacientes con migraña, sus ataques están asociados con déficits neurológicos, que incluyen perturbaciones corticales, denominadas colectivamente aura de migraña(3).
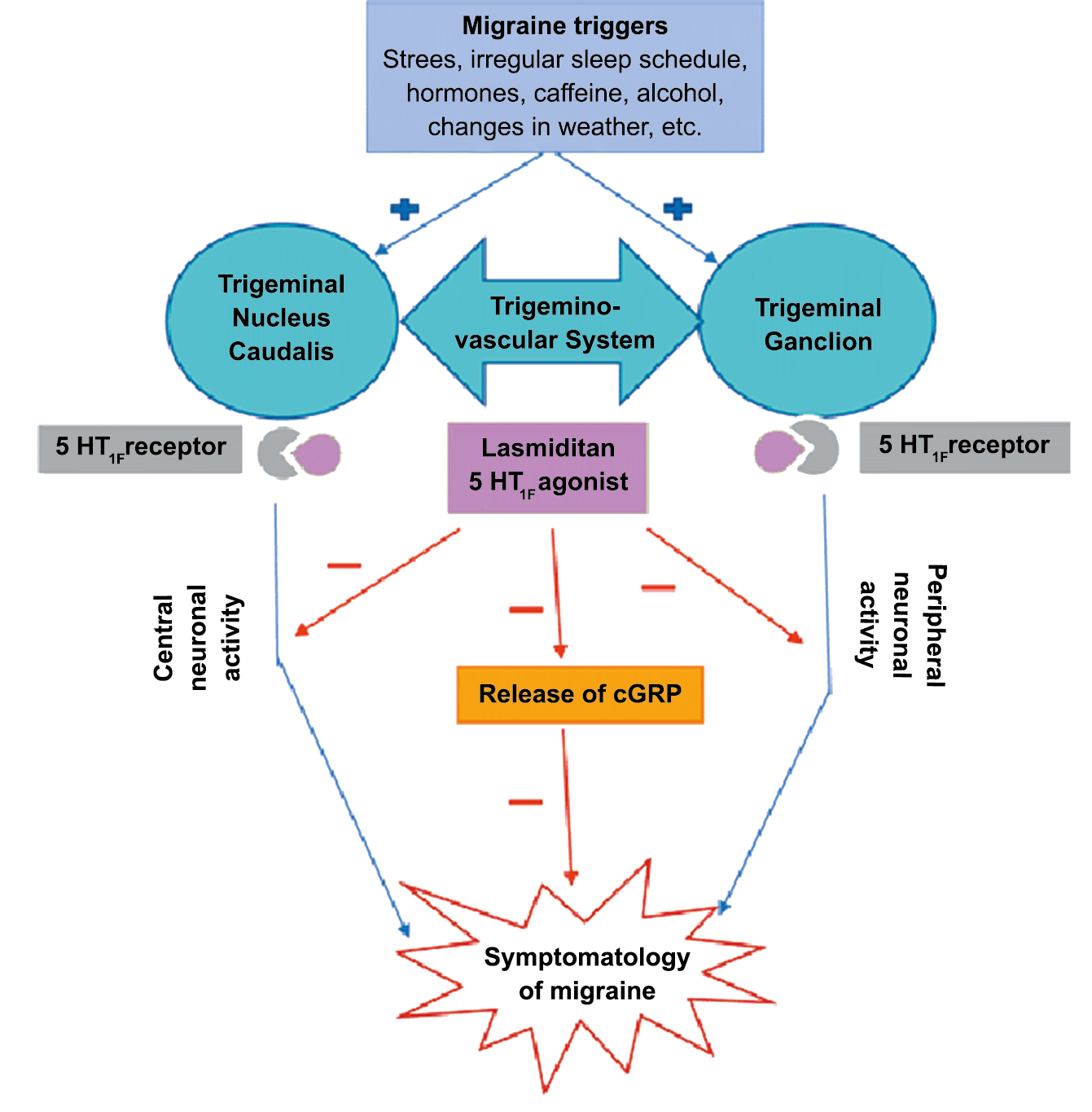
La comprensión de la biología de la migraña a través de una cuidadosa investigación de laboratorio ha llevado al desarrollo de las principales clases de tratamientos: triptanos, agonistas del receptor de serotonina 5-HT1B/1D; gepants, antagonistas del receptor del péptido relacionado con el gen de la calcitonina (CGRP); ditanes, agonistas del receptor 5-HT1F, CGRP anticuerpos monoclonales; moduladores de mGlu5, sin olvidar el efecto ya demostrado por parte de los bloqueantes de los canales de calcio (flunarizina) y de los neuromoduladores; bien se trate de topiramato (Inhibe la acción de la anhidrasa carbónica, bloquea los canales del sodio, aumenta las corrientes de cloro mediadas por GABA, activa las corrientes hiperpolarizantes del K+ e inhibe la activación de receptores a ácido glutámico tipo AMPA), valproato de sodio (Inhibe las enzimas de degradación del GABA y puede aumentar su síntesis, además de tener un papel inhibidor de la transmisión excitatoria de ciertos aminoácidos, entre ellos el glutámico y reducir el umbral de conductancia del calcio y el potasio) y de los antidepresivos de diversas clases como la amitriptilina (antidepresivo tricíclico que impide la recaptación y la inactivación de la noradrenalina y la serotonina en las terminaciones nerviosas) y venlafaxina (inhibe principalmente la recaptación de serotonina y en menor grado de noradrenalina, en la membrana presináptica neuronal, potenciando la neurotransmisión a nivel del sistema nervioso central). El propanolol tiene un efecto antagonista de los adrenoceptores β1 en las neuronas del núcleo VPM (postero medial ventral) del tálamo que responden a la entrada nociceptiva trigeminovascular. Se ha demostrado que el propanolol previene los cambios en el comportamiento y el flujo sanguíneo cerebral inducido por la depresión cortical propagada, además de bloquear los canales de sodio del cerebro(103), modulación del sistema catecolaminérgico central(104) probablemente a través de receptores β-adrenérgicos centrales e interacción cruzada con receptores de serotonina(105) (Figura 4).
Aspectos históricos
El dolor de cabeza se conoce desde hace probablemente 6000 años, aunque no está claro cuál tipo. La migraña, de hecho, en su construcción más moderna del tipo crónico, puede reconocerse fácilmente en el trabajo de Willis en el siglo XVII(4). El debate sobre la fisiopatología de la migraña se ha centrado en gran medida en los mecanismos neurales o vasculares que están implicados en el desencadenamiento y la conducción de los ataques. Los argumentos han existido durante varios siglos y han estado en alternancia. Históricamente, hace casi 150 años ambos parecían ser frecuentes. Edward Livinging pensaba que la migraña era una enfermedad del cerebro. En su famoso texto “Sobre la migraña, enfermedades del dolor de cabeza y algunos desórdenes aliados: Una contribución a la patología de la tormenta nerviosa”, describe la migraña como el resultado de una “tormenta nerviosa, una tendencia heredada a la descarga de la fuerza nerviosa, una “convulsión neuronal”(5). Al mismo tiempo, Peter Wallwork Latham en su publicación “On Nervous or Sick-Headache”(6), describió el origen probable de la migraña a través de la vasodilatación provocada, curiosamente, por el aura. Luego a partir de 1940 Wolff y colaboradores revisaron la idea de que la migraña probablemente fuera un trastorno vascular(7). Este concepto persistió durante casi cinco décadas hasta que el desarrollo de sumatriptán lo apoyó(8) y, paradójicamente, llevó a cuestionarlo(8), pues se demostró que el sumatriptán tenía efectos tanto vasculares como neurales, se desarrolló entonces la teoría de que el dolor en la migraña se debía a una inflamación estéril de las meninges durales causada por la activación antidrómica del nervio trigémino(9).
Situación actual
La migraña definitivamente involucra el cerebro. En algunos aspectos, la controversia sigue siendo bastante similar y gira en torno a dos aspectos: la iniciación y el origen del dolor.
Aunque el inicio de una crisis dolorosa se asocia con frecuencia con desencadenantes internos y externos, este concepto ya está siendo revisado por una interpretación más central que surge cuando se considera la fase premonitoria. El origen de los mecanismos neuronales que subyacen a la afección primaria en personas susceptibles, es decir, ¿qué desencadena el dolor? Aún no se conoce. Sin duda, está ampliamente aceptado que la migraña implica la activación y sensibilización de las vías trigémino vasculares, así como del tronco encefálico y los núcleos diencefálicos(10). Se ha sugerido que la migraña puede considerarse como un estado cerebral de excitabilidad alterada(11). Ciertamente, los síntomas premonitorios de la migraña pueden ocurrir muchos días antes del dolor de cabeza y son síntomas neurológicos de naturaleza no nociceptiva, lo que apunta a un origen en el cerebro. Además, se han identificado múltiples genes responsables de la migraña hemipléjica familiar (FHM, por sus siglas en inglés) y se ha descubierto una predisposición genética en estudios familiares. Estos elementos, entre otros, brindan un fuerte apoyo al hecho que los migrañosos pueden estar genéticamente predispuestos o ser susceptibles
TEORÍAS DE LA MIGRAÑA
Las teorías de la migraña han cambiado mucho a lo largo de los años, a menudo cerrando el círculo y volviendo a las teorías originales de hace 250 años. Sin embargo, en los últimos 60 a 70 años se ha visto el mayor desarrollo de teorías como consecuencia de la investigación médica y del avance de las tecnologías científicas. Desde las observaciones de Harold Wolff y sus colegas en la década de 1940, donde la estimulación mecánica y la distensión de los vasos sanguíneos craneales resultaron en dolor de cabeza en pacientes sometidos a cirugía craneal, una “teoría vascular de la migraña” tomó impulso. Sin embargo en contraste con la suposición común, las pulsaciones experimentadas durante un ataque de migraña no están sincronizadas con las contracciones cardíacas ya que las pulsaciones dolorosas tienen una frecuencia más baja(93).
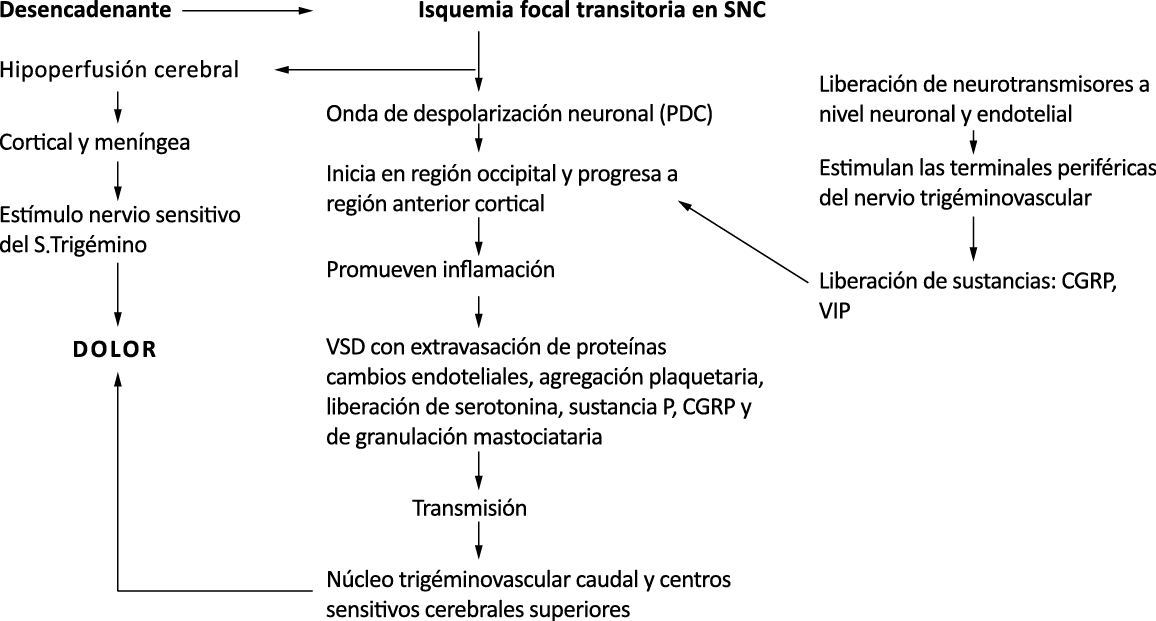
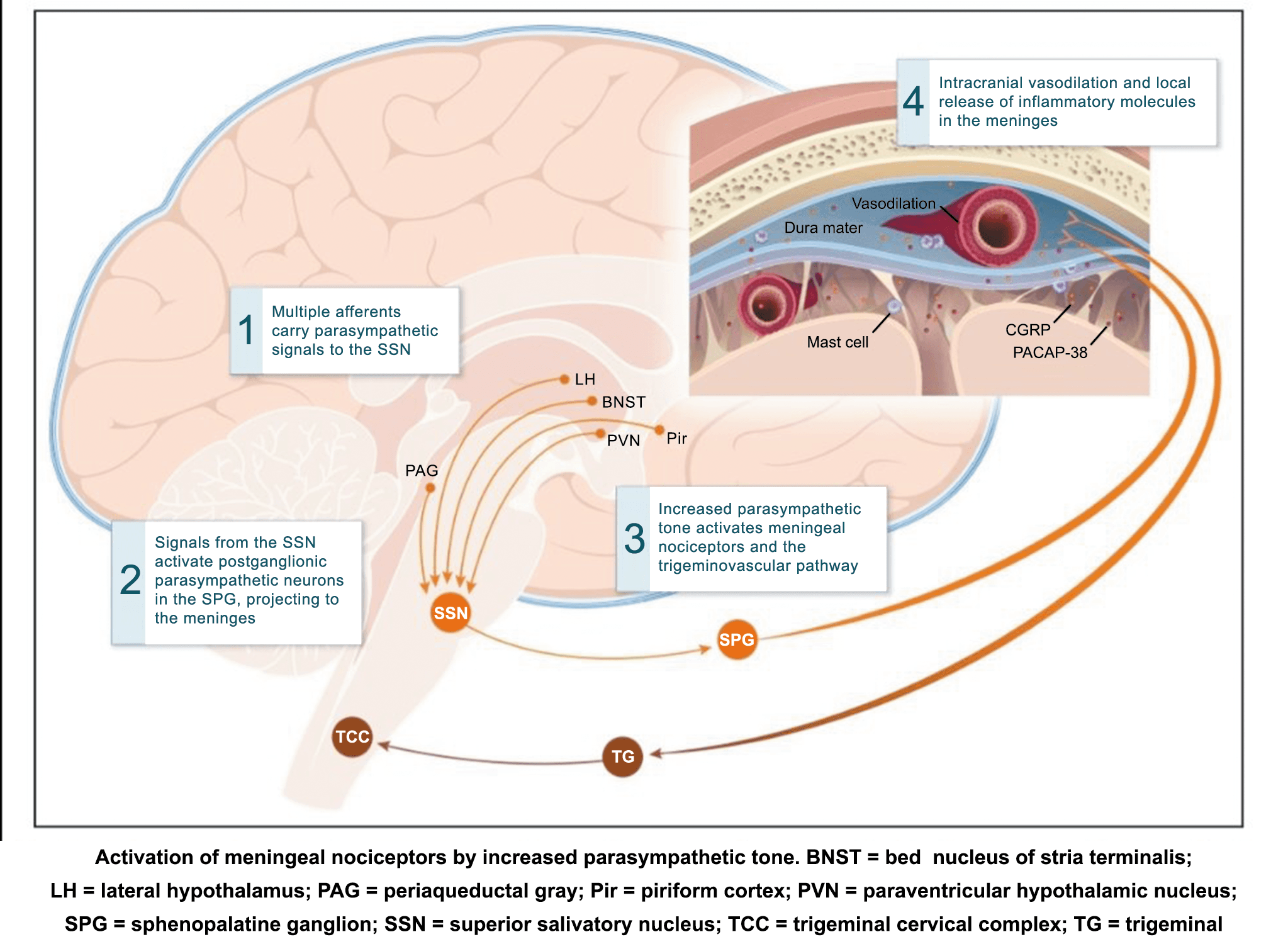
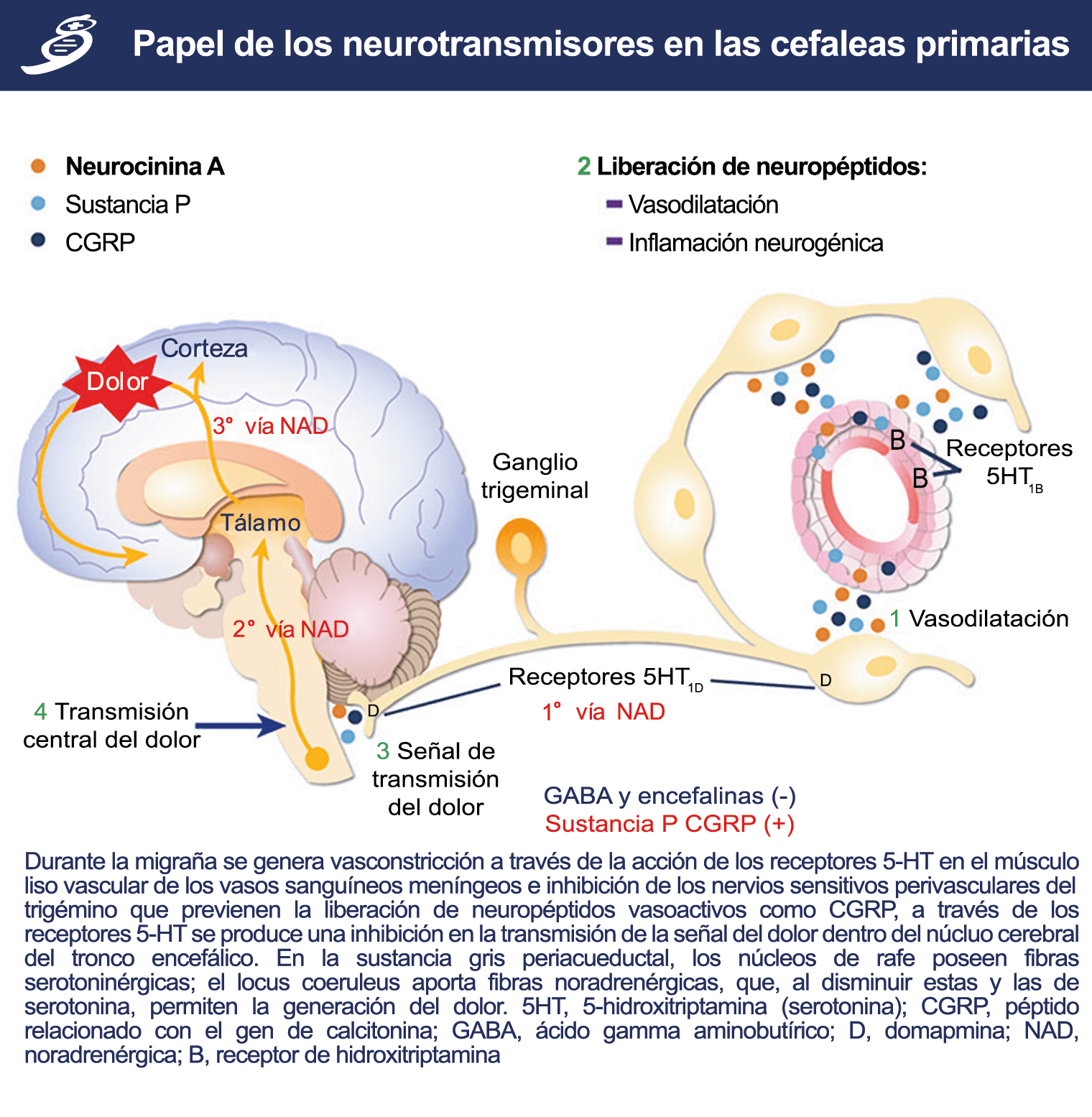
La importancia de la vasculatura intracraneal, o su inervación de fibras nerviosas, sigue siendo significativa. Los estudios de Ray y Wolff(94) demostraron que la estimulación de la duramadre, particularmente alrededor de los vasos sanguíneos durales y cerebrales, puede generar dolor similar al de la cefalea, además de náuseas, mientras que la estimulación lejos de los vasos fue ineficaz. Como se describió anteriormente, los vasos sanguíneos de la duramadre están ricamente inervados por axones nociceptivos no mielinizados (fibras C) y mielinizados (fibras Aδ) que se originan en el ganglio del trigémino y contienen neuropéptidos vasoactivos (serotonina, dopamina, histamina, péptido relacionado con el gel de la calcitonina). Estos datos consolidan la teoría de que la cefalea en la migraña está mediada por la activación de las fibras nerviosas nociceptivas que inervan los vasos sanguíneos meníngeos. La pregunta clave es qué hace que estas fibras se activen durante la migraña. Una propuesta en la década de 1980 fue que una “inflamación neurogénica estéril” de las meninges durales puede resultar en la activación de la inervación perivascular para desencadenar la migraña(95). La activación está impulsada por la liberación dural local de mediadores inflamatorios endógenos como CGRP, sustancia P, neuroquinina A y prostaglandinas, que aumentan el flujo sanguíneo local (predominantemente impulsado por CGRP), la fuga de proteínas plasmáticas de los vasos sanguíneos, la desgranulación de mastocitos y la agregación de las plaquetas.
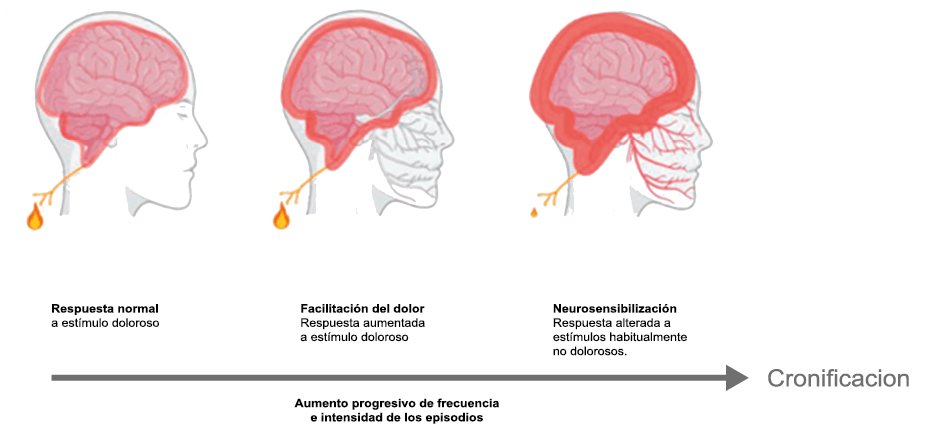
Hoy en día, la migraña se describe más comúnmente como un “trastorno neurovascular” y específicamente como un trastorno del cerebro. Una teoría que ha cobrado importancia en los últimos 15 años, que sostiene este punto de vista y ha servido para explicar muchas características de la migraña que antes no se habían explorado o simplemente no estaban claras es la “sensibilización periférica y central”. La sensibilización es una característica común de muchos trastornos de dolor crónico y se supone que también está presente durante la migraña. Es una activación y sensibilización de larga duración de los nociceptores periféricos y las neuronas nociceptivas centrales y puede explicar la longevidad del ataque de migraña y la transición hacia la migraña crónica, así como síntomas relacionados específicos(96). La sensibilización central de las neuronas trigeminovasculares ha ayudado a nuestra comprensión de la base neural de la hipersensibilidad extracraneal en la migraña, la derivación del dolor y la alodinia a las áreas faciales periorbitarias y cutáneas(97). Es probable que la sensibilización de las neuronas trigeminotalámicas explique la alodinia cutánea generalizada en las regiones extracefálicas(98).
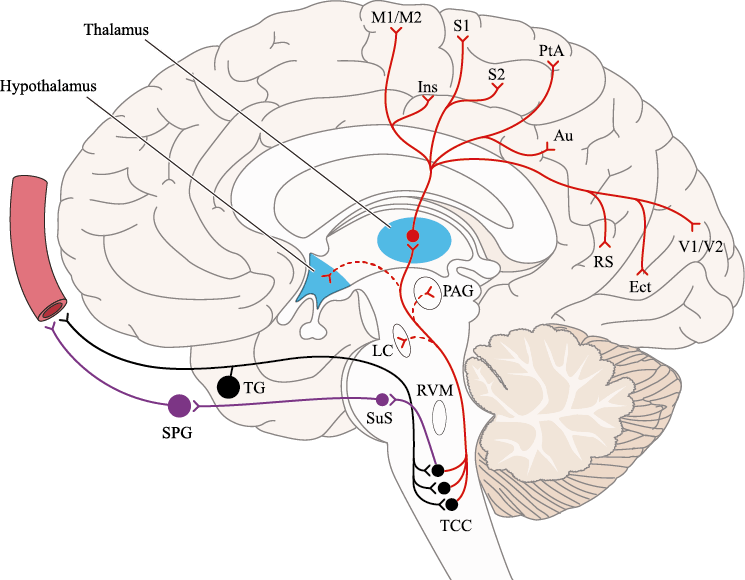
Un principio importante de esta teoría es que el desencadenante principal de la migraña proviene de la periferia, en los vasos sanguíneos de la duramadre, con la activación del sistema trigeminovascular proveniente del disparo de las neuronas trigeminales periféricas de primer orden en respuesta a una lesión nociceptiva o señales provenientes de las meninges que liberan mediadores neuroinflamatorios. La activación sostenida de las neuronas nociceptivas meníngeas durales provoca la activación secuencial y la sensibilización de las neuronas trigeminovasculares de primer orden (nociceptores periféricos), segundo orden (a nivel del TCC-complejo trigémino cervical) y tercer orden (trigeminotalámico), (Figura 5) así como la activación ascendente del cerebro, tallo y otras estructuras diencefálicas(97).
Se cree que esta activación secuencial explica la naturaleza pulsátil del dolor en la migraña, la hipersensibilidad sensorial nociceptiva, los síntomas neurológicos asociados que incluyen náuseas, vómitos, alteración de la alimentación y el sueño, así como la alteración cognitiva. Clínicamente, sin embargo, hay muchas lagunas. Los síntomas premonitorios en la migraña pueden estar presentes 24 a 48 h antes del dolor de cabeza y están representados por cambios en la activación en las regiones del cerebro medio e hipotalámico. Los desencadenantes de la migraña, como la privación de sueño y alimentos y el estrés, están bajo control homeostático.
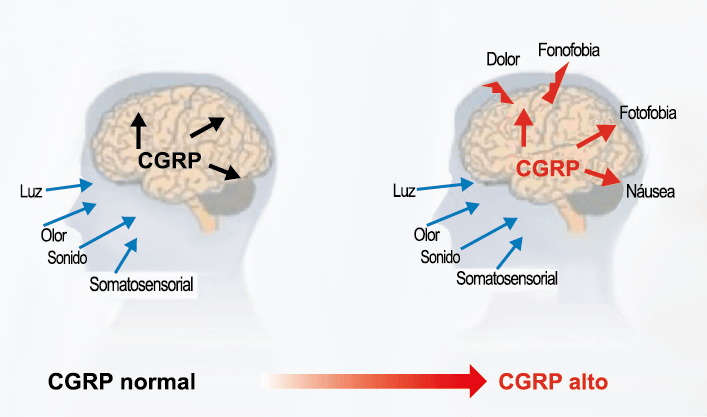
Ha existido una fuerte creencia de que la CSD, el correlato experimental del aura, es el principal evento desencadenante de la activación meníngea (la evidencia a favor y en contra se discutió anteriormente). Como se mencionó, el aura solo está presente en aproximadamente un tercio de los migrañosos, aunque es un sello común en las formas raras identificadas genéticamente. Hay pruebas contradictorias de excitabilidad cortical en la migraña; por un lado, las personas con migraña son más susceptibles a la activación visual y tienen una mayor sensibilidad a tales estímulos. Esto puede resultar de hiperexcitabilidad, particularmente en migraña con aura(99), con un umbral reducido para producir fosfenos(100). Sin embargo, existen informes similares de estados de hipoexcitabilidad y umbrales de fosfeno significativamente más altos(101). Parecería más razonable suponer que existe un cambio cortical alterado entre estados hipo e hiperexcitables que da como resultado una desregulación de la función cortical normal. Alternativamente, gran parte de la evidencia que respalda la hiperexcitabilidad puede resultar de alteraciones trigeminovasculares de orden inferior a través de las redes talamocorticales, como la participación talámica recientemente identificada en la fotofobia. (Figura 6)
Si el desencadenante de la migraña no proviene de la periferia o de la depresión de extensión cortical, ¿cómo se explica la provocación de un ataque de migraña?
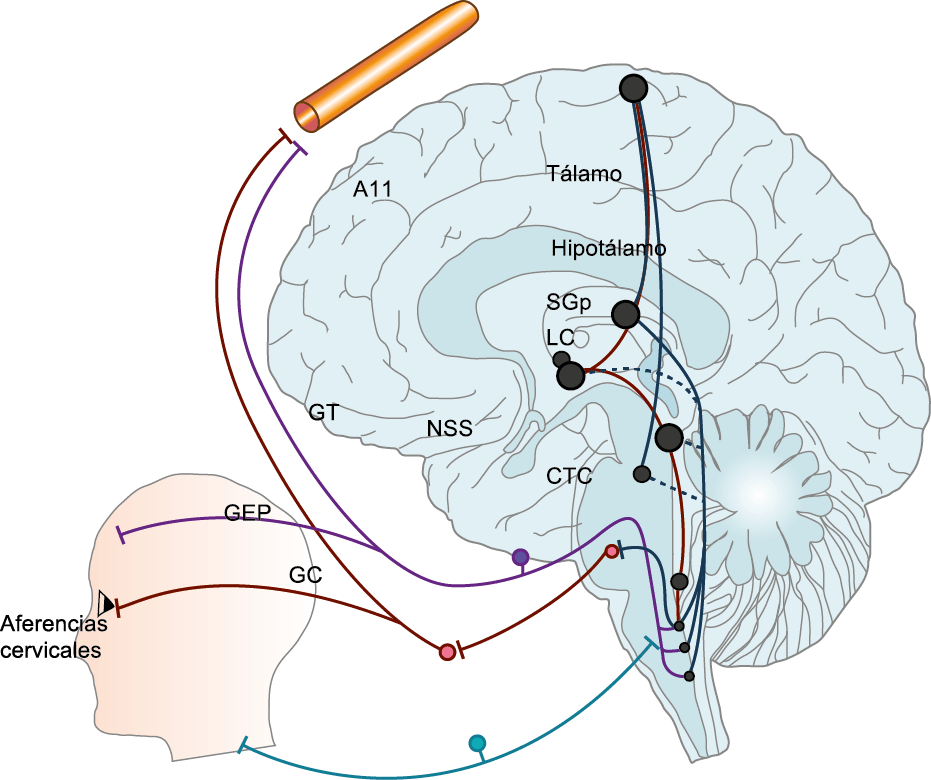
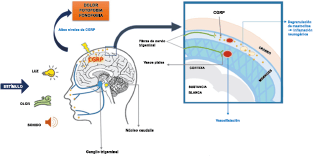
El dolor de la migraña es, sin duda, una consecuencia de la activación o la percepción de la activación de los mecanismos neurovasculares, de allí que sea considerado un trastorno puramente neuronal. Mucha evidencia indica que los cerebros de las personas con migraña pueden ser diferentes en la forma en la cual responden a la estimulación sensorial, incluso interictalmente(102). Una hipótesis alternativa y que lo abarca todo es que el cerebro está en el centro del desencadenamiento de la migraña. Más que una activación secuencial de diferentes regiones del cerebro, la migraña es un trastorno del cerebro y por lo tanto, se considera un “estado cerebral”, que es una consecuencia de cambios o disfunciones en las regiones del tronco encefálico y el hipotálamo, que contribuyen a su vez a cambios a nivel celular y vascular en muchas regiones del cerebro. Esta hipótesis establece que la migraña puede describirse mejor como una consecuencia de la disfunción en el tronco encefálico y los núcleos hipotalámicos que normalmente modulan o activan las entradas sensoriales, como el tacto, la luz, los sonidos y los olores. Estos núcleos del tronco encefálico y del hipotálamo pueden considerarse “mediadores de la migraña” y su disfunción puede llevar al fracaso de los mecanismos de integración y filtrado cerebrales, lo que da como resultado la percepción de activación de los sistemas sensoriales en condiciones normales. La compleja red de conexiones entre las regiones del tronco encefálico, que incluyen PAG, RVM, locus ceruleus y SuS y los núcleos diencefálicos, incluidos el hipotálamo, el tálamo y la corteza, pueden conducir a la generación de síntomas a través de la misma disfunción central. La disfunción en estas regiones, a través del control descendente del tráfico nociceptivo trigeminovascular, puede conducir a la percepción del dolor de cabeza a través de la palpitación de los vasos normales y la disfunción continua puede conducir a la sensibilización central de las neuronas trigeminovasculares y la exacerbación del dolor con la actividad física normal, así como en la piel: alodinia cefálica y extracefálica. La convergencia de entradas sensoriales en el tálamo que se proyectan a la corteza puede explicar la hipersensibilidad a la luz, los sonidos y los olores. La misma disfunción puede conducir a cambios homeostáticos, controlados por el hipotálamo, relacionados con el sueño, la alimentación y la actividad. La alteración general de la función cortical y subcortical puede desencadenar eventos como el aura migrañosa y extenderse a una incapacidad general para funcionar correctamente. Los factores genéticos heredados juegan claramente un papel en la predisposición a la susceptibilidad a la migraña, al igual que el papel de los desencadenantes potenciales de la migraña, cuyo vínculo común parece jugar en el corazón de la homeostasis cerebral en el hipotálamo y el tronco encefálico. (figura 7).
Siendo un trastorno cerebral complejo y multifacético que en su totalidad puede durar varios días, se ha dividido clásicamente en cuatro fases: premonitoria, aura, cefalea y postdrómica. Estos pueden ocurrir en un orden secuencial lineal, pero en la mayoría de los casos, las fases de la migraña muestran una significativa superposición, de modo que el orden lineal es a la vez atractivo y engañoso en su simplicidad. Es importante recalcar que algunos síntomas, como el cansancio o el deterioro de la concentración, bien pueden presentarse en todas las fases. En realidad, solo el dolor de cabeza, por su ausencia o presencia, es quien se destaca, aunque, como se verá, es solo parte del cortejo sintomático.
Síntomas premonitorios
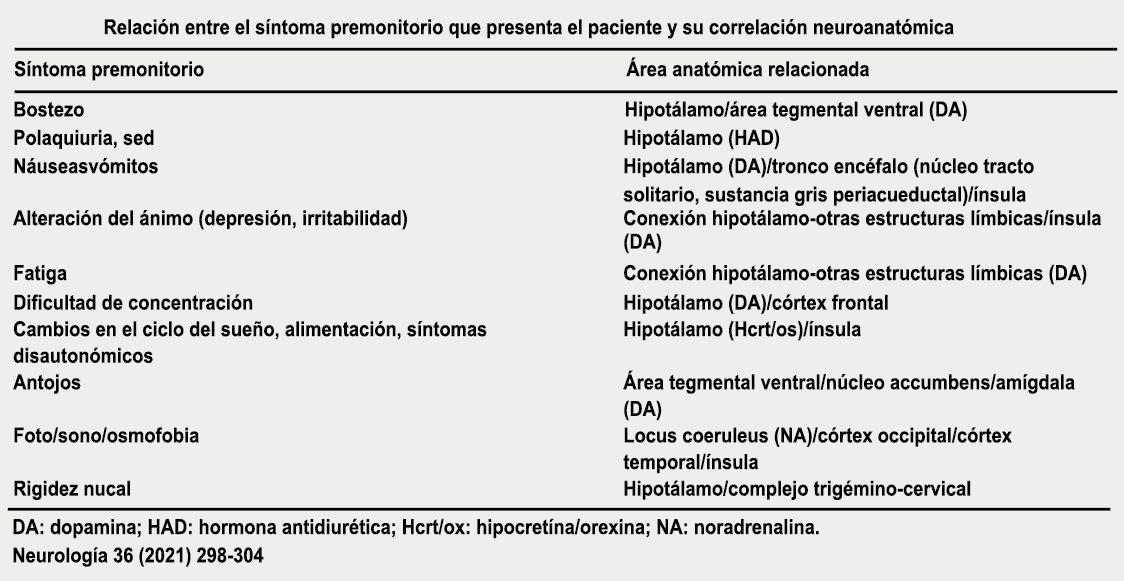
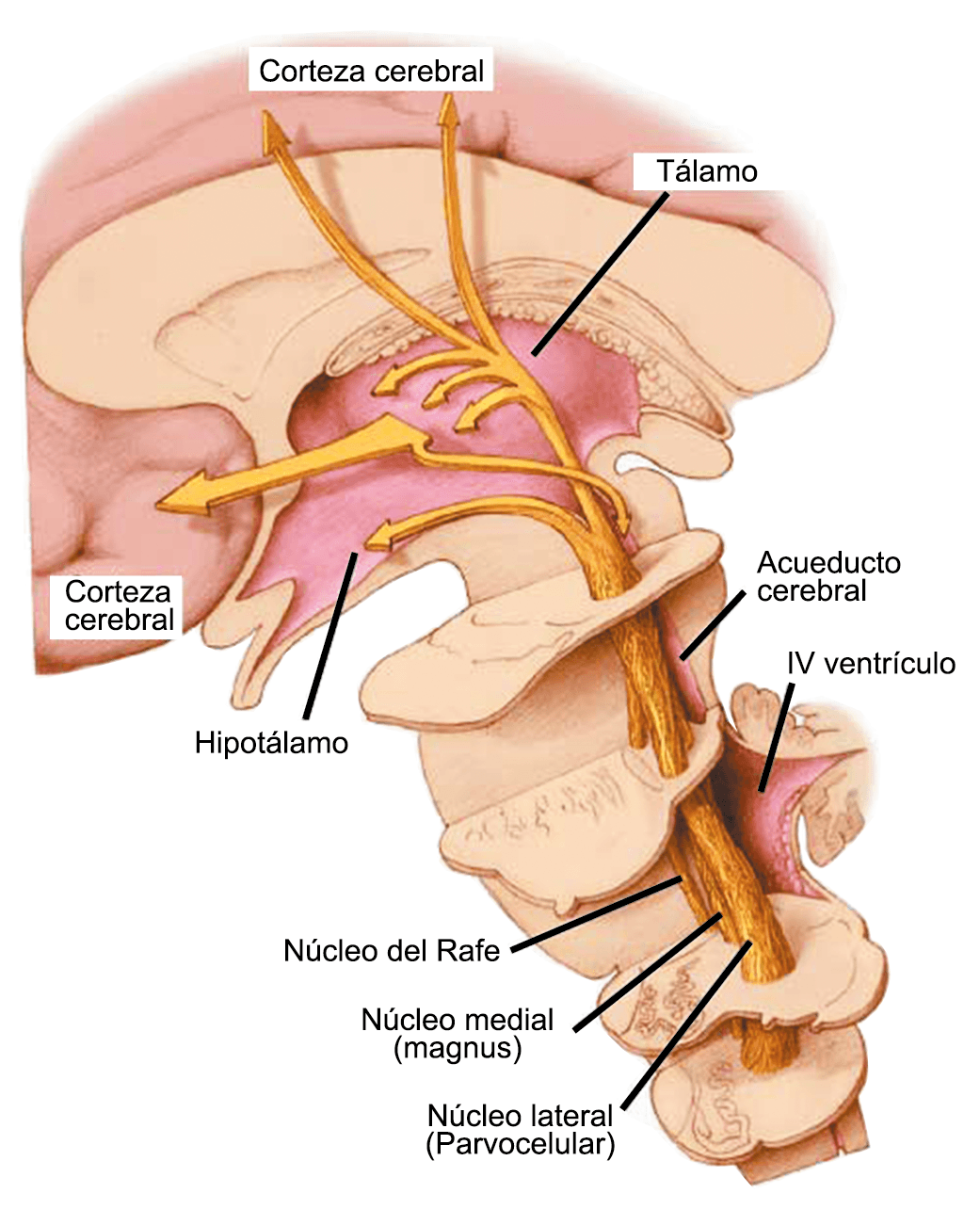
Los síntomas premonitorios son parte del cuadro clínico que acompaña a los dolores de cabeza en las crisis de migraña. Por definición, ocurren antes del inicio del dolor y son diferentes del aura de la migraña. Los síntomas premonitorios más frecuentemente reportados fueron sensación de cansancio y agotamiento (72%), problemas de concentración (51%) y rigidez de nuca (50%). Menos comunes, pero de manera importante, asociados con una mayor previsibilidad de los ataques de migraña fueron los bostezos, la visión borrosa, la sensibilidad al ruido, las dificultades de la función cortical superior (como leer, escribir, hablar, pensar) y sentirse emocional e irritable. Además, la regulación homeostática parece estar alterada durante la fase premonitoria que se manifiesta con sed, náuseas, hambre, micción frecuente o estreñimiento(36). Este patrón complejo de síntomas ha llevado a la hipótesis de que los síntomas podrían ser de naturaleza hipotalámica y/o cerebrales difusos(37). La mayoría de los migrañosos los experimentan mucho antes de que se inicie la típica migraña, pero que ya le indican al paciente que un dolor de cabeza está en camino. Estas señales premonitorias pueden preceder a la fase de dolor de cabeza hasta 72 horas, también se han informado cambios en el estado de ánimo y la actividad, irritabilidad, fatiga, antojos de alimentos, bostezos repetitivos, rigidez en el cuello y fonofobia. Pueden perdurar hasta bien entrada el aura, la cefalea e incluso las fases posdrómicas. La consistencia de estos síntomas permite a algunos migrañosos predecir de forma fiable sus crisis dolorosas. El hecho de que estos síntomas sean en gran medida de origen hipotalámico, han sido demostrados por estudios de imagen mediante PET con H2O(12).
Curiosamente, muchos de los factores desencadenantes descritos por los migrañosos, como por ejemplo la falta de sueño, el hambre o la luz brillante, pueden representar síntomas premonitorios de un ataque que ya está en curso. (Tabla 1)
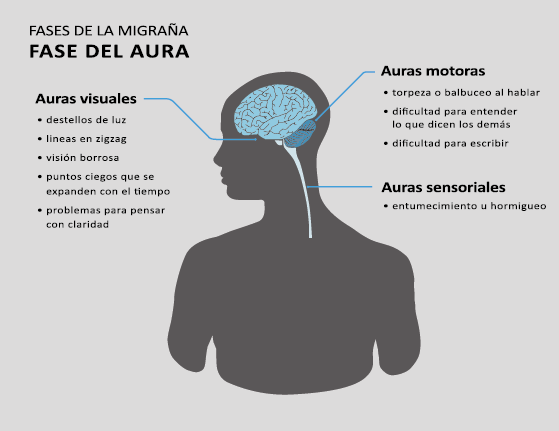
Fase de aura
Alrededor de un tercio de los migrañosos experimentan déficits neurológicos transitorios: el aura de la migraña, en el contexto de sus episodios migrañosos(13). La Clasificación Internacional de la Cefalea 3 Beta, define el aura migrañosa como uno o más déficits neurológicos transitorios, completamente reversibles, de los cuales al menos uno debe tener una localización unilateral, que se desarrollan durante 5 minutos o más y cada déficit dura entre 5 y 60 minutos.
El 26% de los pacientes tiene al menos una de tres auras que dura más de una hora. La forma en que los enfermos suelen lidiar con su migraña es una clara indicación de su hipersensibilidad a la estimulación sensorial. Algunos informarán que durante un ataque se irán a la cama, apagarán las luces y evitarán cualquier tipo de estimulación sensorial de luz, sonido, tacto u olor, como si estuvieran cerrando todas las entradas sensoriales posibles. De hecho, en la clasificación de la migraña se incluye que acompañando la migraña debe haber al menos fotofobia, fonofobia, náuseas y vómitos(38). La fotofobia es uno de los síntomas claves no relacionados con el dolor de cabeza que definen la migraña. Los pacientes generalmente describen que la luz es demasiado brillante (sensibilidad anormal a la luz) o incluso dolorosa al causar o empeorar el dolor de cabeza o los ojos (alodinia fotótica). Incluso en los períodos intercríticos toleran menos luminosidad que los controles sanos y activan la corteza visual (cuneus y giro lingual) cuando se exponen a diferentes intensidades luminosas. La fotofobia en la migraña puede tomar la forma de alodinia fótica, donde la luz en sí es desagradable sin dolor, o hay hipersensibilidad fotótica. Puede mostrar fenómenos positivos (espectros de fortificación), negativos (escotoma), o ambos. Se encuentra en más del 90% de los casos, generalmente comienza antes de la fase de dolor, pero también puede ocurrir al mismo tiempo o incluso independientemente de cualquier dolor de cabeza. Otras auras descritas son el aura sensorial, motora, del habla, del tronco encefálico y retiniana. Demarquay et al.(19) evaluaron el procesamiento olfativo en migrañosos con hipersensibilidad olfativa habitual y encontraron que durante la estimulación del olor, los migrañosos habían aumentado el flujo sanguíneo cerebral en el polo temporal izquierdo. Por lo tanto, algunos síntomas específicos, como la hipersensibilidad olfativa, se asocian con una respuesta cortical única incluso fuera de los ataques. Es muy común la superposición de las fases del aura y del dolor de cabeza. En los que padecen síntomas de aura motora, como en la migraña hemipléjica, los síntomas de aura suelen mostrar una duración más prolongada de hasta 72 horas. En conjunto, estos datos sugieren que varias áreas corticales y subcorticales en el cerebro de las personas con migraña responden de manera diferente a los estímulos externos en comparación con los controles sanos. Esto podría implicar que el cerebro de una persona con migraña es “hiperexitable”(20) con anomalías funcionales precondicionadas que se exacerban concertadamente durante los ataques de migraña. Sin embargo, se debe mantener una mente abierta y evaluar la presencia de factores de riesgo cardiometabólico conocidos o desconocidos por el paciente, con el fin de considerar los diagnósticos diferenciales. (Figura 8)
Se cree que una onda transitoria de despolarización neuronal de la corteza (Figura 9), la depresión de propagación cortical (CSD), es el mecanismo cerebral fisiopatológico subyacente al fenómeno clínico del aura de la migraña. Leão(21) estableció un mecanismo subyacente hipotético. Luego de haber estimulado eléctricamente la corteza de conejo y encontrar una depresión EEG que se propagaba a una velocidad similar de 3 mm/min centrífugamente desde el sitio de estimulación sugirió que podría ser la base del aura migrañosa. Esta hipótesis está fundamentada en la correlación entre las características neurofisiológicas de una CSD, su propagación retinotópica en la corteza visual y las características y dinámicas de los déficits visuales(14, 15) y en las observaciones indirectas derivadas de los estudios de imágenes que respaldan aún más este concepto(16) Sin embargo, sobre la base de la comprensión actual de la migraña, es poco probable que la CSD esté involucrada en el inicio del síndrome completo de la migraña(17).
Fase de dolor de cabeza
De acuerdo con la Clasificación Internacional de la Cefalea, el dolor de cabeza en la migraña debe tener al menos dos propiedades que incluyen: unilateral, de calidad pulsátil, de intensidad moderada o severa y agravamiento lo que lleva a evitar la actividad física de rutina. La activación de las vías del dolor trigeminovascular que inervan la vasculatura dural es responsable de esta cualidad de dolor de cabeza. En la última edición de la Clasificación Internacional de la cefalea, la migraña se define como episodios de dolor de cabeza que duran de 4 a 72 horas y se acompañan de náuseas, fotofobia y fonofobia, o ambas. La cefalea se caracteriza por ser unilateral, pulsátil, de intensidad moderada o severa y agravada por la actividad física; dos de estas características son suficientes para cumplir los criterios diagnósticos. Se distingue la migraña crónica, que ocurre 15 o más días al mes, de la migraña episódica, que puede durar 30 minutos a varios días. Típicamente, comienzan varias horas después de despertar y empeoran a medida que progresa el día. Pocas veces despiertan a los pacientes del sueño.
La migraña se ha asociado con el hipometabolismo de las áreas centrales de procesamiento del dolor, incluida la ínsula bilateral, la corteza cingulada anterior y posterior bilateral, la corteza premotora y prefrontal izquierda y la corteza somatosensorial primaria izquierda, lo que sugiere una disfunción del procesamiento del dolor central en el estado interictal que posiblemente refleja la preparación del cerebro para desarrollar ataques de migraña.
La relevancia del tronco encefálico para la fisiopatología de la migraña está respaldada por estudios clínicos e investigaciones básicas. En presencia de la crisis dolorosa, se ha demostrado que hubo un aumento del flujo sanguíneo cerebral en el mesencéfalo, la protuberancia rostral dorsal cerca del PAG y los núcleos del rafe(22) y además se localizaron varios síntomas de migraña en diferentes áreas del cerebro concomitantemente con la experiencia de dolor de cabeza en la corteza cingulada, fotofobia en la corteza de asociación visual y fonofobia en el auditivo, corteza de asociación. Estas señales desaparecieron después de la terminación exitosa del ataque. Sin embargo, el aumento de flujo sanguíneo cerebral en el tronco encefálico persistió en la fase temprana sin dolor. Por lo tanto, esta estructura refleja no solo un síntoma de migraña, sino que también indica una disfunción de importancia para la generación o el mantenimiento del ataque de migraña en sí.
De forma interictal en los migrañosos, la estimulación luminosa activa la corteza visual de forma bilateral. Durante la migraña espontánea, la baja luminiscencia no provoca activación cortical visual pero lo hace durante la crisis dolorosa aunque esta activación fue estadísticamente menor que durante la migraña. Se ha demostrado que, durante la crisis y en los periodos intercríticos, hay hiperexcitabilidad cortical a la luz. Incluso esta presente después del alivio del dolor.
Fase posdrómica
La fase posdrómica ha estado descuidada, debido en parte a los resultados de los estudios que se han centrado en esta última etapa de un ataque de migraña, ellos indican que sus síntomas característicos reflejan los observados durante el período premonitorio(18). Los síntomas típicos posteriores al síndrome incluyen: cansancio, dificultades para concentrarse y rigidez en el cuello. No está claro si estos síntomas se inician en la fase premonitoria y persisten a lo largo de etapa de cefalea hasta llegar a esta fase, pues también pueden iniciarse durante la fase de cefalea o incluso aparecer después de que finaliza la fase dolorosa. Los pacientes migrañosos comúnmente relacionan los síntomas de esta fase con la medicación que eliminó con éxito su dolor de cabeza, lo que indica que estos síntomas aparecen o reaparecen después de que la fase de dolor de cabeza ha terminado.
En el cerebro de las personas con migraña se activan distintas áreas, las cuales desempeñan roles diferentes dentro del complejo sintomático ya sea desencadenando el ataque, generando el dolor o participando en algunos de los síntomas neurológicos asociados que ocurren durante un ataque. Desde el punto de vista fisiológico y fisiopatológico se ha aprendido sobre la asociación de estas diferentes regiones cerebrales entre sí, definiendo la participación de núcleos diencefálicos, del tronco encefálico y áreas corticales. También se ha podido determinar cómo se procesa la información nociceptiva de las estructuras craneovasculares, lo que da como resultado la percepción del dolor de cabeza durante la migraña, así como los síntomas sensoriales neurológicos asociados.
Fisiopatología de la cefalea
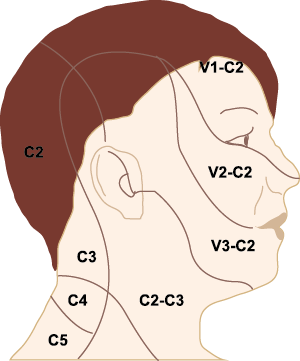
El dolor de cabeza asociado con un ataque de migraña, puede incluir la región frontal, temporal, parietal, occipital y cervical alta, es consecuencia de la activación del sistema trigeminovascular. (Figura 10)
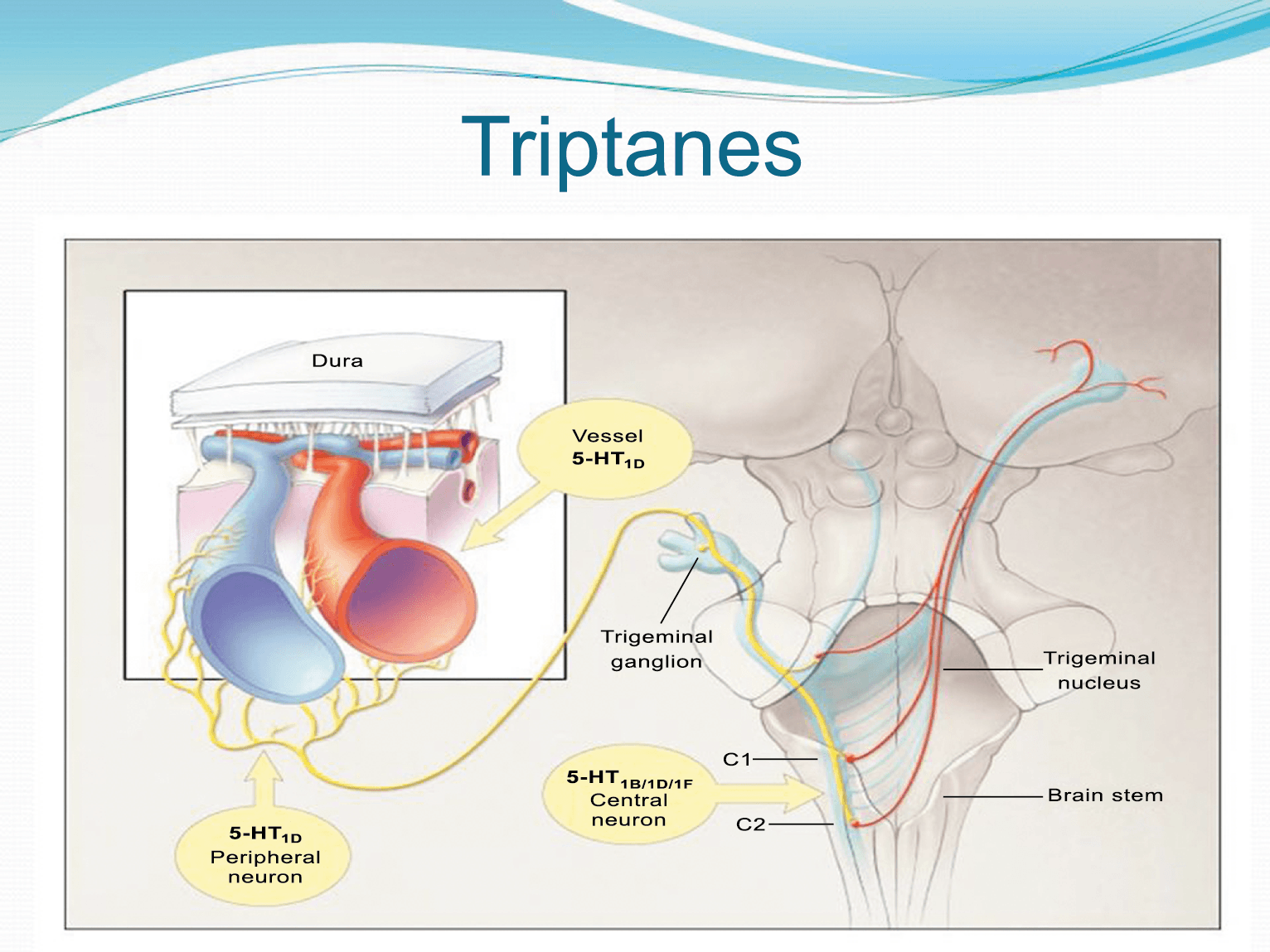
La anatomía del sistema trigéminovascular ha sido bien descrita durante los últimos 70 años y esto ha ayudado a comprender la fisiopatología de la migraña y la distribución de su dolor. Se sabe que el cerebro es en gran parte insensible, pero un rico plexo de fibras nerviosas nociceptivas que se originan en el ganglio del trigémino inervan los vasos sanguí- neos de la piamadre, la aracnoides y la duramadre, incluidos el seno sagital superior y la arteria meníngea media, así como las grandes arterias cerebrales. (Figura 11) (23)
La inervación nociceptiva de la vasculatura intracraneal y las meninges incluye proyecciones axonales no mielinizadas (fibras C) y mielinizadas delgadas (fibras Aδ), principalmente a través de la división oftálmica (V1) del nervio trigémino, pero también, en menor medida, a través del nervio maxilar. (V2) y divisiones mandibulares (V3). También hay inervación neuronal de la duramadre desde los ganglios de la raíz dorsal cervical (24) (Figura 12).
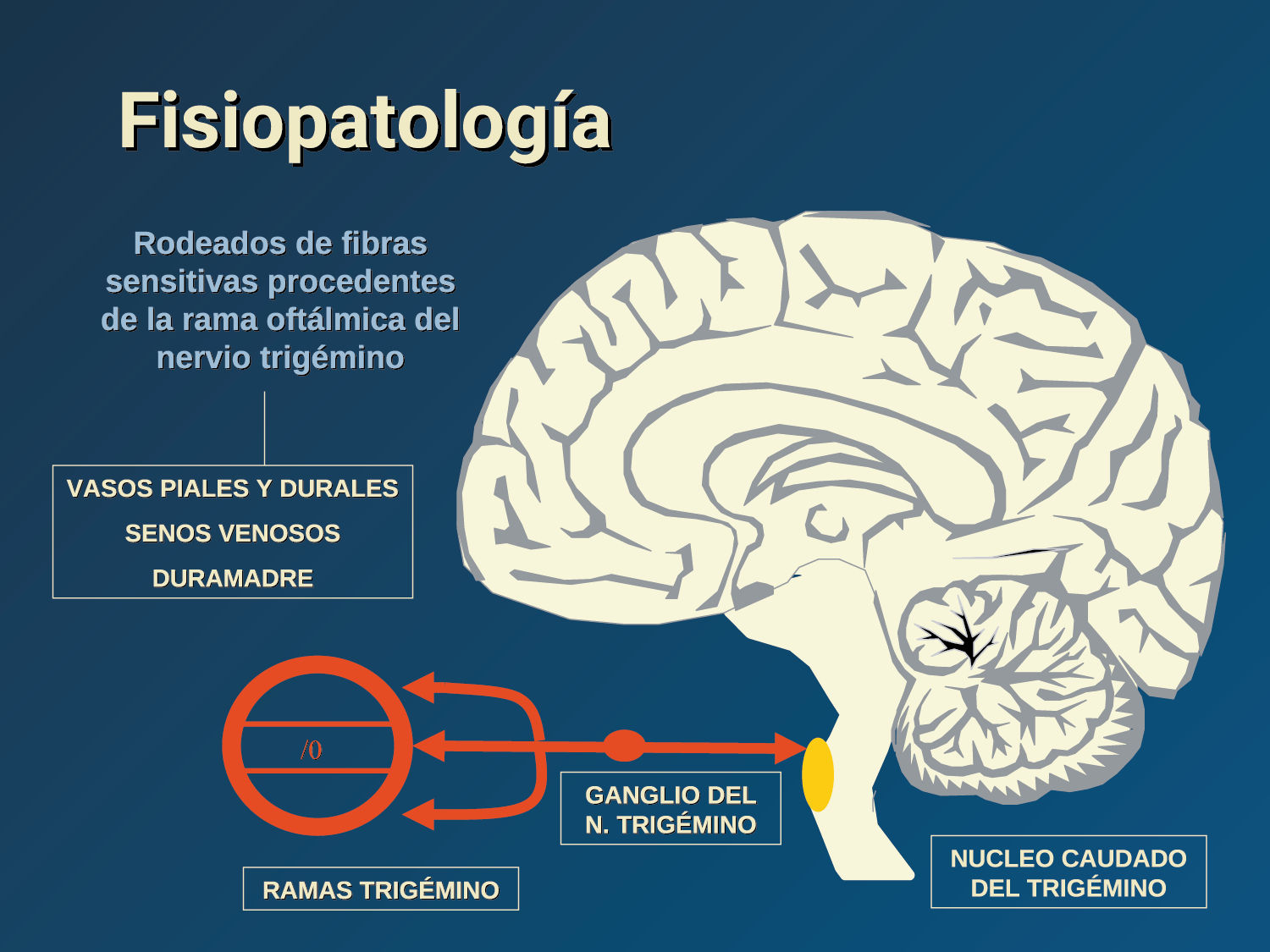
Los terminales axónicos de las fibras nerviosas nociceptivas que inervan la duramadre contienen neuropéptidos vasoactivos CGRP, sustancia P, neurocinina A y péptido activador de la adenilato ciclasa pituitaria (PACAP)(25) (Figuras 13 y 14), que se cree que son liberados tras la estimulación que causa la vasodilatación de los vasos durales y piales(26) (Figura 15).
Hay una proyección aferente central desde el ganglio del trigémino que ingresa a la médula caudal del tronco encefálico, a través del tracto del trigémino, que termina en el núcleo espinal del trigémino caudalis (TNC), así como la médula espinal cervical superior (C1–C2). (Figura 16)
Las fibras nociceptivas Aδ y C terminan predominantemente en las láminas superficiales, I y IIo, así como en las láminas más profundas V–VI del TNC y extensión cervical. La estimulación de la vasculatura dural en modelos animales, incluidos los senos transverso y sagital superior y la arteria meníngea media, da como resultado la activación de las neuronas en las regiones TNC, C1 y C2 de la médula espinal cervical, conocidas en conjunto como el complejo trigeminocervical (TCC).
Estos datos sugieren que el núcleo del trigémino se extiende hasta el asta dorsal de la región cervical superior en un continuo funcional que incluye la extensión cervical, ello explica la distribución de la percepción del dolor en la migraña sobre las regiones frontal y temporal, además de la participación de parietal, occipital y regiones cervicales superiores(27).
Por lo tanto, se cree que la naturaleza intensa y pulsátil del dolor en la migraña es el resultado de la activación, o la percepción de activación, de estas entradas nociceptivas de las estructuras intracraneales y extracraneales, que convergen y se transmiten a través del TCC. (Figura 17)
Toda la información nociceptiva de las estructuras craneovasculares se transmite a través del TCC y mediante conexiones ascendentes a otras áreas del tronco encefálico y el diencéfalo, involucradas en el procesamiento del dolor y otra información sensorial. La activación de la vía antes mencionada contribuye a la percepción del dolor durante la migraña y también a los síntomas autonómicos, endocrinos, cognitivos y afectivos que duran todo el episodio migrañoso.
El procesamiento del dolor es complejo y está mediado por una red de estructuras neuronales que incluyen la corteza cingulada, las ínsulas y el tálamo(28). El tálamo está en el corazón del procesamiento central y la integración de la información nociceptiva y se considera un centro de retransmisión para manejar la información sensorial entrante e incluso modularla. Se cree que la llamada “matriz del dolor”, que incluye el tálamo, así como las áreas somatosensoriales primarias (S1) y secundarias (S2), la corteza cingulada anterior (CCA) y la corteza prefrontal, está involucrada en la integración de todos los sentidos, respuestas afectivas y cognitivas al dolor y se activan durante el procesamiento nociceptivo(28). (Figura 18)
Siendo el tálamo el centro principal para el procesamiento de la información nociceptiva sensorial en el cerebro, la transmisión de esta información para su procesamiento en las estructuras corticales donde los individuos la perciben es un factor clave. (Figura 18) Los tratamientos agudos como los “triptanos” (agonistas de los receptores 5-HT1B/1D)(29) y los antagonistas de los receptores CGRP(30) pueden inhibir las entradas nociceptivas durales agudas. Del mismo modo, los preventivos de la migraña propranolol(31), valproato de sodio(32) y topiramato(33)también pueden inhibir las entradas trigeminotalámicas nociceptivas durales en el VPM (Núcleo ventral posteromedial).
La evidencia de la activación talámica durante la migraña es clara(34) y su papel va más allá del mero relevo en el procesamiento de información nociceptiva sensorial a las cortezas somatosensoriales.
La presencia de alodinia cutánea
La percepción del dolor en respuesta a estímulos normalmente inocuos, tiene dos formas de presentación y responden cada una a mecanismos diferentes. La alodinia en las regiones cefálica, es consecuencia de la sensibilización periférica y de las neuronas trigeminales y explica la hipersensibilidad cutánea facial en la región del dolor referido experimentada por muchos migrañosos(35). Esta alodinia, similar a la hipersensibilidad a la luz y al sonido que experimenta el paciente y que puede o no acompañarse de hiperalgesia, ocurren en hasta dos tercios de los pacientes con migraña(39). La alodinia ha sido reconocida al menos desde el siglo XIX(40). En el paciente con migraña se manifiesta predominantemente en la región cefálica/facial y el dolor puede ser provocado simplemente cepillando o secando el cabello, afeitándose o duchándose. La alodinia extracefálica se extiende a otras regiones convertirse en una alodinia en todo el cuerpo, como cuando se viste(41) y es explicada por la sensibilización central de las neuronas trigeminotalámicas. Curiosamente, la frecuencia y severidad de la alodinia cutánea es más marcada en la migraña más frecuente y severa y puede considerarse un marcador de la transformación en formas más severas y crónicas de migraña(42). (Figura 19)
DESENCADENANTES DE LA MIGRAÑA
Los ataques de migraña generalmente se consideran de naturaleza espontánea y que pueden ocurrir sin ningún factor desencadenante o precipitante tangible. Sin embargo, muchos pacientes con frecuencia informan que hay circunstancias exógenas o endógenas conocidas a las que le atribuyen de manera confiable sus ataques de migraña, o al menos aumentan la probabilidad de que ocurran.(45) Tal vez esta confusión y la clara falta de claridad acerca de qué elementos pueden estar involucrados en la precipitación de los ataques de migraña, es consecuencia de dos hechos principales. En primer lugar, es probable que los pacientes no entiendan bien el fenotipo completo y la duración del síndrome de migraña, pero a veces tampoco los médicos. El ataque de migraña no comienza con la percepción del dolor de cabeza, o en el caso del aura, durante los primeros déficits neurológicos del aura, sino muy probablemente de 24 a 48 horas antes de estos síntomas, durante la fase premonitoria(46). Tal y como se describió en el subtítulo Fase Premonitoria , muchos pacientes informan tener la sensación de que se avecina una migraña aunque no sientan dolor varios días antes y esto a menudo se caracteriza por síntomas inespecíficos, como cansancio, falta de atención inapetencia. De hecho, los estudios de imágenes han demostrado que hay cambios significativos en la activación cerebral mientras se perciben estos síntomas(47). Es posible que la difuminación de las líneas entre los síntomas premonitorios y los desencadenantes de la migraña haya llevado a los pacientes y a los médicos a creer que ciertos factores ambientales que se cree que precipitan su ataque de migraña en realidad representan un síntoma de su síndrome, como el antojo de ciertos alimentos, incluidos el chocolate y queso, así como cansancio. (Figura 20)
“Interrupción del sueño”
La relación entre la migraña y el sueño se ha debatido durante varios años(48). El sueño presenta una dualidad: se considera un medio eficaz para aliviar el dolor asociado con la migraña por una parte y por la otra, la migraña puede surgir durante el sueño nocturno o después de un breve período de sueño diurno y los ataques pueden estar precedidos por la falta de sueño. Además, la somnolencia también puede surgir durante varias fases de un ataque de migraña(49).
Muchos pacientes también describen la privación del sueño o el sueño interrumpido o reducido continuamente, como un posible desencadenante de la migraña(50). La regulación del sueño, particularmente dentro del hipotálamo, está fuertemente ligada al desencadenamiento de la migraña. Los núcleos del mesencéfalo están involucrados al desencadenamiento de la migraña, además de contribuir a los síntomas asociados a través de proyecciones al hipotálamo y otros núcleos del tronco encefálico y diencefálico. Además de controlar la modulación del dolor, las cé- lulas “ON: ENCENDIDAS” y “Off: APAGADAS” en RVM (médula medial rostro-ventral) y NRM (nú- cleos del rafe magno) (Figura 21).
También están involucradas en el control de respuestas a estímulos externos inocuos, actividad motora y procesos homeostáticos(51). Las células “On: ENCENDIDAS” están activas dependiendo del estado durante las horas de vigilia, pero no durante la alimentación y la micción, o durante el sueño, mientras que las células “Off: APAGADAS” están activas durante el sueño y activas durante la vigilia solo antes de micción, así como durante la alimentación. Por lo tanto, la activación de células “Off: APAGADAS”, durante el sueño, la alimentación y la micción, evita que respondan a estímulos inocuos e incluso nocivos agudos, mientras que durante la vigilia la activación de células “On: ENCENDIDAS” facilita el estado de alerta a los estímulos sensoriales. Además, la vlPAG (Área ventrolateral de la sustancia gris periacueductal) (Figura 22) participa en los mecanismos de excitación, a través de la activación de las neuronas dopaminérgicas durante la vigilia y la inactivación de estas neuronas durante el sueño. En el contexto de los posibles desencadenantes de la migraña, la privación del sueño es un proceso homeostático que da como resultado respuestas neuronales específicas en la vía PAG-RVM, en particular, la activación de las células “ON” y la inhibición de la activación de las células “OFF”.
En los migrañosos, es posible que las respuestas alteradas de las células “ON” y “OFF” a los cambios nociceptivos y homeostáticos, como la interrupción del sueño y las proyecciones neuronales de los sistemas PAG-RVM a los núcleos hipotalámicos y las neuronas nociceptivas espinales y del trigémino, descritas anteriormente, contribuye a desencadenar los síntomas sensoriales y fisiológicos de la migraña.
FARMACOLOGÍA DE LA MIGRAÑA: TRATAMIENTOS
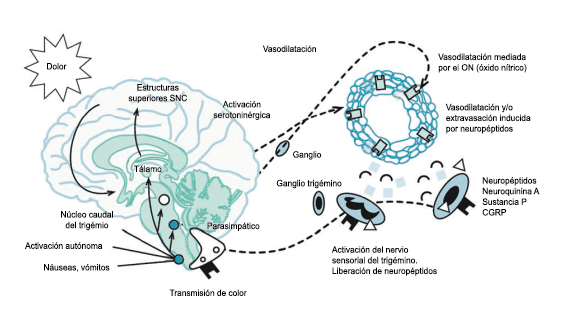
La activación del sistema trigeminovascular da como resultado la liberación de varios neuropéptidos basados en las inervaciones simpática, parasimpática y sensorial de la vasculatura craneal, que se resumen en la figura 23.
La inervación simpática se caracteriza por NPY y norepinefrina(52), ambos de los cuales son vasoconstrictores, mientras que las fibras parasimpáticas se caracterizan por VIP y PACAP, que se encuentran entre los vasodilatadores más potentes(24).
Las inervaciones sensoriales se caracterizan por la sustancia P, CGRP y PACAP (53).
La serotonina
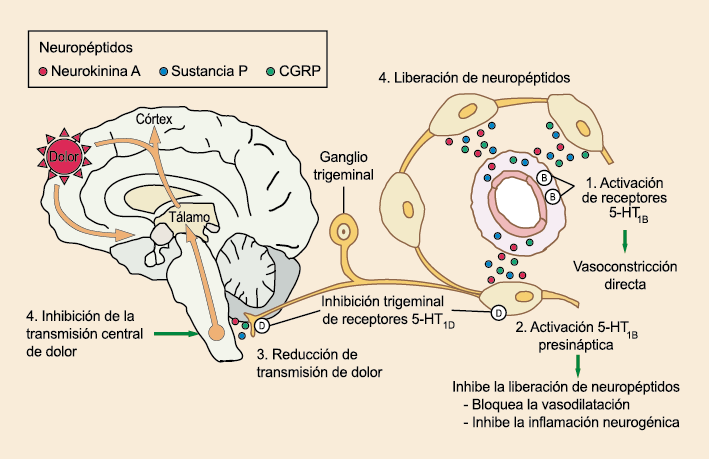
La participación inicial de la serotonina (5-HT) en la migraña fue postulada hace más de 50 años.(54) (Figura 24)
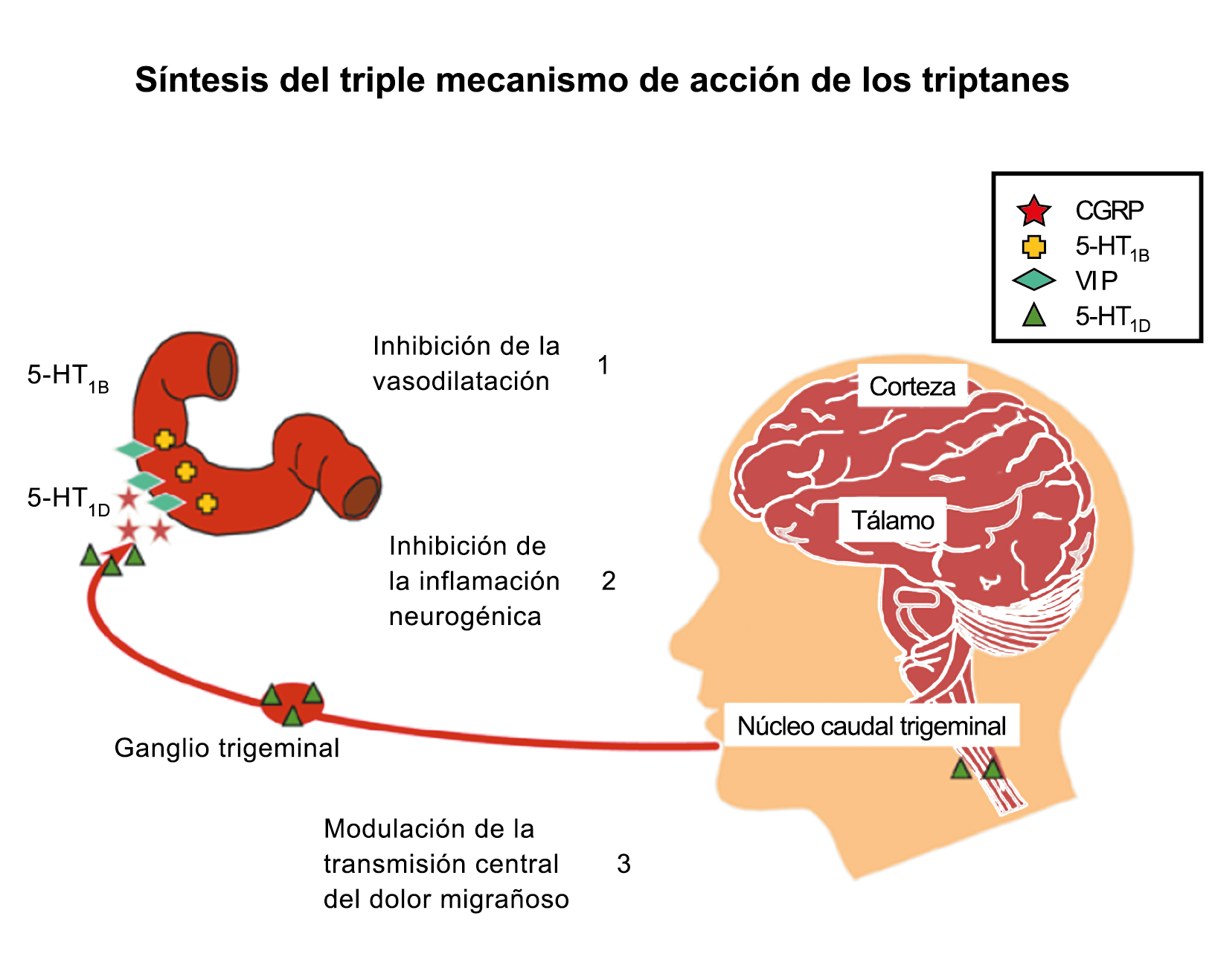
Posteriormente diversos estudios demostraron que la infusión de 5-HT podría abortar tanto la cefalea inducida por reserpina(55) como la espontánea(56). En respuesta a estos estudios, el sistema del receptor 5-HT ganó mucha atención, lo que culminó con el descubrimiento de los triptanos, la serotonina y los agonistas del receptor 5-HT1B/1D(57). Todos exceptuando el 5-HT3 (canal iónico controlado por ligando) son receptores acoplados a proteína G. Si bien los triptanos se clasifican como agonistas de los receptores 5-HT1B/1D, la mayoría también activa en menor medida los receptores 5-HT1A, 5-HT1E o 5-HT1F(58). Originalmente, los triptanos se desarrollaron para actuar sobre la vasculatura craneal, una idea apoyada por sus claras acciones vasoconstrictoras(59) (Figura 25) y la expresión preferencial del receptor 5-HT1B en los vasos craneales más que en los periféricos(60).
Un mecanismo neural de los triptanos en las terminaciones del nervio trigémino periférico podría inhibir la liberación de neuropéptidos proinflamatorios y la vasodilatación dural neurogénica(63), lo cual era consistente con el tema en desarrollo de la migraña como una enfermedad neurovascular. (Figuras 26 y 27)
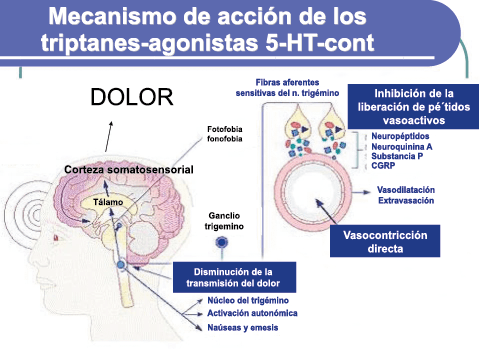
En la figura 28 se muestra un resumen de los fármacos y sus mecanismos de acción en el contexto del sistema trigémino vascular.
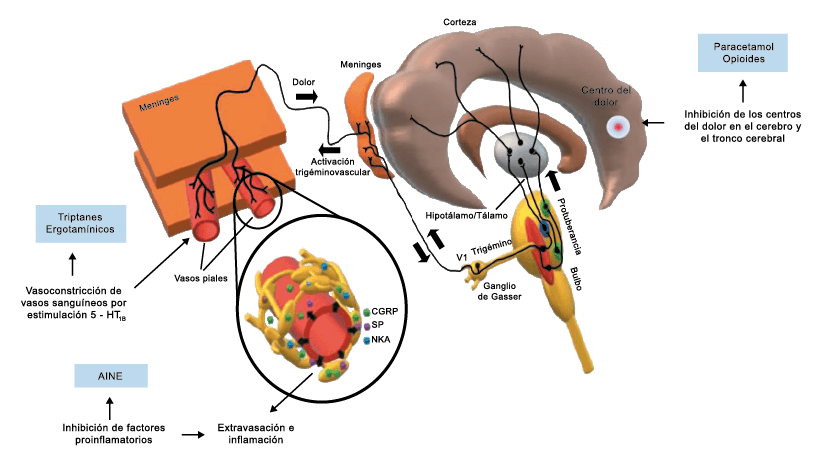
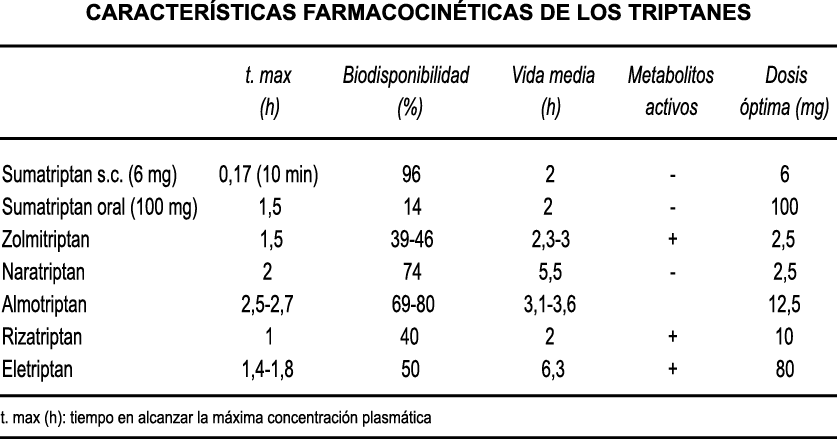
A pesar de la clara farmacología de los triptanos en el tratamiento de la migraña, su uso es limitado en pacientes con factores de riesgo cardíaco(61), lo que obliga a dirigirse a receptores que carecen de acción vasoconstrictora. Una de esas dianas es el receptor 5-HT1F, que es activado hasta cierto punto por algunos triptanos, como el naratriptán y no por otros, como el rizatriptán(62). (Tabla 2)
Toxina botulínica
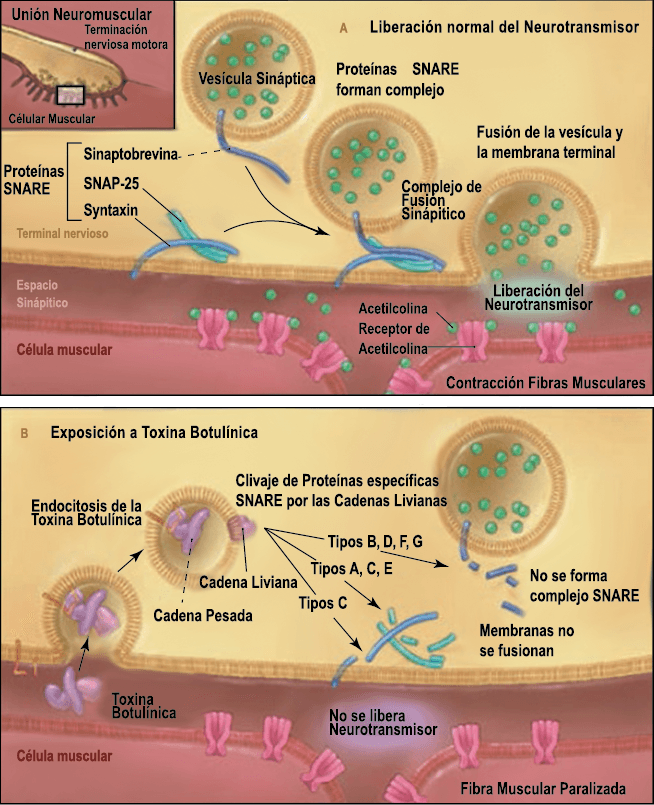
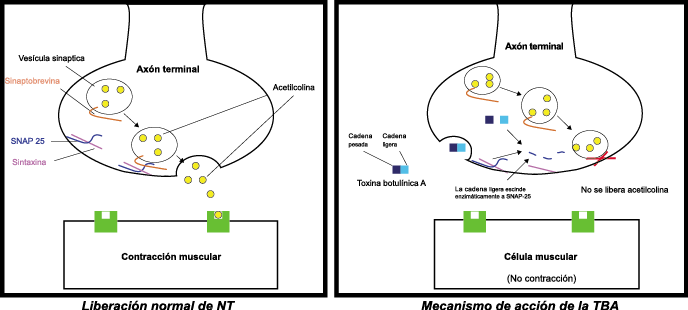
La toxina botulínica tipo A (BT-A) fue descrita por primera vez por Binder y colaboradores en 1991 como tratamiento preventivo para la migraña al observar que cuando pretendían reducir líneas de expresión faciales en algunos pacientes estos vieron como también se redujeron los dolores de cabeza por migrañas. La BT-A ya ha sido utilizada previamente como tratamiento de otras afecciones tales como hiperhidrosis axilar, estrabismo, acalasia o alteraciones musculares como distonías y espasticidad. La inyección vía subcutánea de toxina botulínica tipo A (BT-A) en el cuero cabelludo, fue aprobada en 2011 por la FDA. Diversos estudios in vitro e in vivo han demostrado que la TBA inhibe la liberación de la sustancia P y de aminoácidos excitadores y péptido relacionado con el gen de la calcitonina) (Figura 29). Más recientemente, su efecto antinociceptivo ha sido relacionado con inhibición de liberación de glutamato (Figura 30).
Existen dos tipos de toxina botulínica comercializados: la tipo A y la tipo B, con características farmacológicas distintas. La toxina botulínica tipo A posee el mayor número de estudios llevados a cabo en el campo de la cefalea. Los numerosos estudios abiertos y la experiencia clínica positiva con la toxina difieren de los resultados en estudios rigurosos controlados, los cuales se han llevado a cabo en varios tipos de cefaleas primarias, como la migraña episódica, crónica, cefalea tensional, mixta y cervicogénica. Existe evidencia reciente(84) que indica que un subgrupo de pacientes que padece cefalea crónica diaria, la cual tiende a ser de carácter refractario, parece responder a este novedoso tratamiento.
Antagonistas de los canales de calcio
Los antagonistas de los canales de calcio: La flunarizina es un bloqueador selectivo tipo L de la entrada de calcio, disminuye su influjo excesivo a través de la membrana celular. Sin efectos a nivel cardiaco, impide la vasoconstricción producida por la hipoxia a nivel periférico y tiene un modesto efecto estabilizador de las propiedades homorreológicas del eritrocito.
Teniendo en cuenta uno de los efectos iniciadores de la migraña, como la depresión cortical propagada, que probablemente es causada por una alteración de la permeabilidad neuronal y tiene una disfunción posterior de canales iónicos, es importante recalcar la presencia de dos mecanismos neuronales subyacentes: la sensibilización periférica y la sensibilización central(85). Las estructuras sensitivas intracraneanas reciben inervación de ramas del trigémino estimuladas por liberación neuronal y endotelial, abriendo receptores en las terminales neurogénicas. Esto lleva al incremento de la concentración de calcio intracelular, seguido de fosforilación y activación de proteincinasa (PKC) y tirosincinasa (TryK) que, a su vez, por mecanismos moleculares aumenta la liberación de sustancias como la CGRP y VIP y promueve la inflamación neurogénica. Este proceso conduce a la sensibilización periférica dada por la disminución del umbral de respuesta de las fibras meníngeas, potenciándose el dolor y la inflamación(86). Por otra parte Las concentraciones de serotonina, la cual se forma a partir del L-triptófano, son altas en las plaquetas y en el tracto gastrointestinal. Este hecho adquiere crucial importancia ya que la serotonina viaja por el torrente sanguíneo hasta el cerebro donde sufre procesos de hidroxilación y descarboxilación para realizar sus funciones a nivel central(22). Las neuronas ricas en esta sustancia se encuentran alrededor del tallo cerebral y de la formación reticular en especial en el núcleo caudalis del trigémino en donde no solo donde tienen la mayor concentración de serotonina, también estas neuronas predominantemente serotoninérgicas se encuentran en mayor concentración, lo cual es un factor especial para desencadenar el proceso de activación del sistema trigémino vascular en el ataque de migraña. Los receptores especiales de 5HT, al activarse, incrementan la hidrólisis de inositol fosfato y generan un aumento de la concentración de calcio en el proceso migrañoso. Además de la serotonina existen diferentes sustancias involucradas en el desarrollo de la cefalea como los receptores dopaminérgicos y la histamina, que tienen efectos vasculares de vasoconstricción o vasodilatación. Existe una hipersensibilidad dopaminérgica en la migraña y en lo que respecta a la histamina, se ha visto incrementada a nivel plasmático por una posible liberación a partir de los leucocitos o por la activación de células mastocitarias. Aunque la histamina no cruza la barrera hematoencefálica esta actúa a través de receptores H1 que están en el endotelio(88).
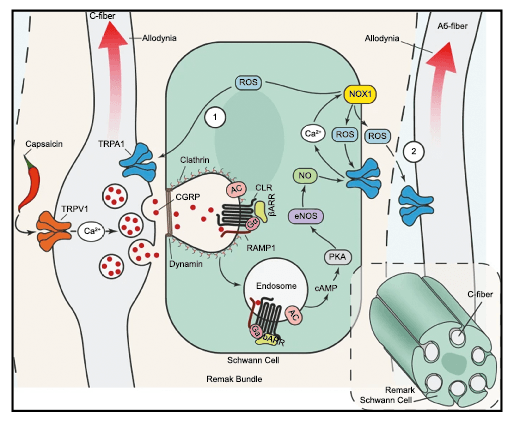
Péptido relacionado con el gel de la calcitonina (CGRP por sus siglas en inglés)
El CGRP (Péptido relacionado con el gel de la calcitonina) es un neuropéptido de 37 aminoácidos con potentes efectos vasodilatadores que se produce mediante empalme alternativo del gen de la calcitonina. Con respecto al sistema nervioso, αCGRP se expresa predominantemente, mientras que βCGRP se asocia con el sistema sensorial entérico(64). El CGRP actúa en dos receptores: uno, el llamado complejo canónico de CGRPreceptor, está formado por un receptor acoplado a proteína G y un único receptor transmembrana que modifica proteína (RAMP) 1(65).
RAMP 1 es fundamental para el transporte y la expresión en la superficie celular del complejo del receptor CGRP y su sobreexpresión da como resultado fenotipos similares a la migraña, incluida la aversión a la luz y la alodinia inducida por CGRP(66) (Figura 32 y 33).
El segundo receptor de CGRP se ha denominado receptor de amilina (AMY1) y consiste en el receptor de calcitonina (CTR) y RAMP 1(67). Ahora se ha demostrado claramente que CGRP activa este receptor(68). La amilina es una hormona glucorreguladora y tiene efectos adicionales en el metabolismo óseo. (Figura 34)
El CGRP está ampliamente distribuido, incluso en el cuerpo estriado, la amígdala, el hipotálamo, el tálamo, el tronco encefálico y el TCC(69). Dentro de las vías trigeminovasculares aferentes primarias, la expresión de CGRP es más alta en el ganglio del trigémino sensorial y sus proyecciones de fibras Aδ y C a los vasos sanguíneos cerebrales y durales, así como centralmente a la médula espinal(70), donde puede afectar proyecciones ascendentes de segundo orden. Se han identificado receptores de CGRP funcionales y sitios de unión en las células del músculo liso de la arteria dural y en los ganglios del trigémino, el tálamo, el hipotálamo, la amígdala, la corteza y el tronco encefálico(71).
El CGRP es un potente vasodilatador que, cuando se administra a personas que padecen migrañas, se sabe que desencadena ataques(72). Se libera durante los ataques espontáneos(73) o provocados(74), que pueden inhibirse mediante el tratamiento con triptanos(75). Además de sus efectos vasculares, CGRP se ha convertido en un modulador clave de la función neuronal, que tiene efectos importantes en los sistemas de neurotransmisores como el sistema glutamatérgico(76). Sobre la base de los datos clínicos(77), se inició un esfuerzo para desarrollar antagonistas de los receptores de CGRP: los “gepantes”: telgacepant demostró una eficacia mejor que el placebo y comparable a los triptanos(78), como el primer antagonista oral del receptor de CGRP con excelente tolerabilidad a largo plazo(79) y también fue bien tolerado en pacientes que estaban siendo investigados por enfermedad arterial coronaria(80). Sin embargo, el desarrollo de telgacepant se detuvo cuando surgieron problemas con las enzimas hepáticas en un estudio preventivo, que resultó bastante positivo(81). Este efecto no se ha informado con rimagepant(82) Los ditanes, tienen como mecanismo el ser agonistas selectivos del receptor 5-hidroxitriptamina tipo 1F (5-HT1F) en la vía trigeminal, hasta ahora ha demostrado no tener efecto vasoconstrictor asociado. Lasmiditán ha sido hasta la fecha de elaboración de esta revisión la primera molécula aprobada por FDA. (Figura 35)
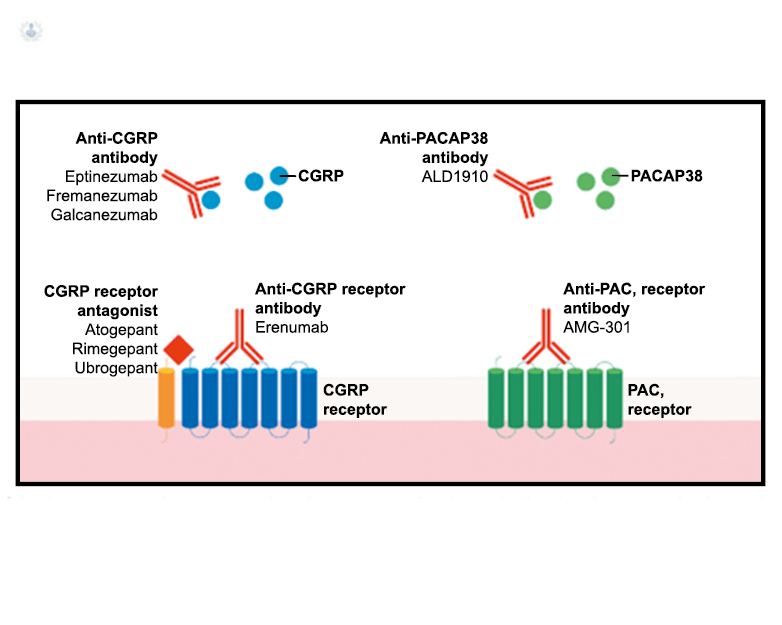
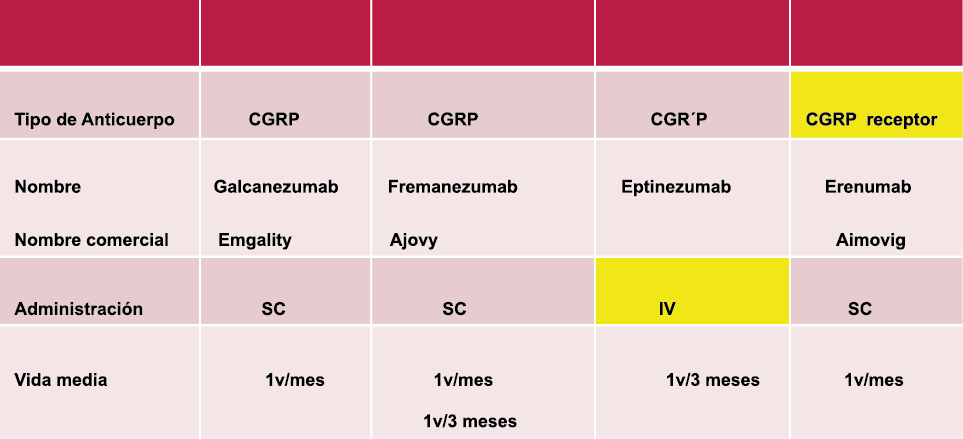
Los anticuerpos monoclonales contra el péptido CGRP y el receptor (numab y nezumabs) se han desarrollado para capitalizar los efectos antimigrañosos comprobados del bloqueo de los mecanismos CGRP como nuevos tratamientos preventivos. Los anticuerpos contra el péptido CGRP, al CGRP y del receptor CGRP (complejo CLR/RAMP1) han presentado datos positivos de ensayos controlados.(83) (Figura 36 y Tabla 3)
El óxido nítrico (NO) es una molécula de señalización gaseosa abundante que, al igual que los neuropéptidos mencionados anteriormente, participa en una variedad de funciones, incluida la vasodilatación dependiente del endotelio(89). Dada la clara función de los donantes de NO en el desencadenamiento de los ataques, no sorprende que hayan recibido una atención similar en modelos preclínicos(90) y activación trigeminovascular (91) y aunque los primeros estudios mostraron resultados beneficiosos(92), no estuvieron exentos de problemas, para el momento de la realización de esta revisión.
Referencias
- Lipton RB, Bigal ME, Diamond M, Freitag F, Reed ML, Stewart WF, Advisory Group AMPP. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology 68: 343–349, 2007.
- Buse DC, Manack A, Serrano D, Turkel C, Lipton RB. Sociodemographic and comorbidity profiles of chronic migraine and episodic migraine sufferers. J Neurol Neurosurg Psychiatry 81: 428–432, 2010
- Rasmussen BK, Olesen J. Migraine with aura and migraine without aura: an epidemiological study. Cephalalgia 12: 221–228, 1992.
- Willis T, Pordage S. Two Discourses Concerning the Soul of Brutes, which Is that of the Vital and Sensitive of Man: The First Is Physiological, Shewing the Nature, Parts, Powers, And Affections Of The Same; And The Other Is Pathological, Which Unfolds the Diseases Which Affect It and Its Primary Seat, to Wit, the Brain and Nervous Stock, and Treats of Their Cures: With Copper Cuts. London: Dring, Harper and Leigh, 1683. [Google Scholar]
- Liveing E. On Megrim, Sick-Headache, and Some Allied Disorders. A Contribution to the Pathology of Nerve-Storms. London: Arts & Boeve Nijmegen, 1873. [Google Scholar]
- Latham PW.On Nervous or Sick-Headache. Cambridge, UK: Deighton, Bell and Co, 1873. [Google Scholar]
- Ray BS, Wolff HG. Experimental studies on headache. Pain sensitive structures of the head and their significance in headache. Arch Surg 41: 813– 856, 1940. [Google Scholar]
- Humphrey PPA, Goadsby PJ. Controversies in headache. The mode of action of sumatriptan is vascular? A debate. Cephalalgia 14: 401–410, 1994.
- Markowitz S, Saito K, Moskowitz MA.Neurogenically mediated leakage of plasma proteins occurs from blood vessels in dura mater but not brain. J Neurosci 7: 4129–4136, 1987.
- Akerman S, Holland P, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nature Rev Neurosci 12: 570–584, 2011.
- Charles A. Migraine: a brain state. Curr Opin Neurol 26: 235–239, 2013.
- Maniyar FH, Sprenger T, Monteith T, Schankin C, Goadsby PJ. Brain activations in the premonitory phase of nitroglycerin triggered migraine attacks. Brain 137: 232–242, 2014.
- Rasmussen BK, Olesen J. Migraine with aura and migraine without aura: an epidemiological study. Cephalalgia 12: 221–228, 1992.
- Charles A. Migraine: a brain state. Curr Opin Neurol 26: 235–239, 2013.
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 31: 17–35, 2011.
- Cutrer FM, Sorensen AG, Weisskoff RM, Ostergaard L, Sanchez del Rio M, Lee J, Rosen BR, Moskowitz MA. Perfusion-weighted imaging defects during spontaneous migrainous aura. Ann Neurol 43: 25–31, 1998.
- Goadsby PJ. Migraine, aura and cortical spreading depression: why are we still talking about it? Ann Neurol 49: 4–6, 2001.
- Blau J. Migraine postdromes: symptoms after attacks. Cephalalgia 11: 229–231, 1991.
- Demarquay G, Royet JP, Mick G, Ryvlin P. Olfactory hypersensitivity in migraineurs: a H(2)(15) O-PET study. Cephalalgia 28: 1069–1080, 2008.
- 750. Stankewitz A, May A. The phenomenon of changes in cortical excitability in migraine is not migraine-specific–a unifying thesis. Pain 145: 14– 17, 2009.
- Leao AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 10: 409–414, 1947.
- Weiller C, May A, Limmroth V, Juptner M, Kaube H, Schayck RV, Coenen HH, Diener HC. Brain stem activation in spontaneous human migraine attacks. Nature Med 1: 658–660, 1995.
- McNaughton FL, Feindel WH. Innervation of intracranial structures: a reappraisal. In:Physiological Aspects of Clinical Neurology edited by Rose FC. Oxford: Blackwell Scientific, 1977, p. 279–293.
- Marfurt CF. The central projections of trigeminal primary afferent neurons in the cat as determined by the tranganglionic transport of horseradish peroxidase. J Comp Neurol 203: 785–798, 1981
- Edvinsson L, Brodin E, Jansen I, Uddman R. Neurokinin A in cerebral vessels: characterization, localization and effects in vitro. Regul Pept 20: 181– 197, 1988.
- Ebersberger A, Averbeck B, Messlinger K, Reeh PW. Release of substance P, calcitonin gene-related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neuroscience 89: 901–907, 1999.
- Bartsch T, Goadsby PJ. Anatomy and physiology of pain referral in primary and cervicogenic headache disorders. Headache Currents 2: 42–48, 2005.
- Derbyshire SWG, Jones AKP, Gyulai F, Clark S, Townsend D, Firestone LL. Pain processing during three levels of noxious stimulation produces differential patterns of central activity. Pain 73: 431–445, 1997.
- Shields KG, Goadsby PJ. Serotonin receptors modulate trigeminovascular responses in ventroposteromedial nucleus of thalamus: a migraine target? Neurobiol Dise 23: 491–501, 2006.
- Summ O, Charbit AR, Andreou AP, Goadsby PJ. Modulation of nocioceptive transmission with calcitonin gene-related peptide receptor antagonists in the thalamus. Brain 133: 2540–2548, 2010.
- Shields KG, Goadsby PJ. Propranolol modulates trigeminovascular responses in thalamic ventroposteromedial nucleus: a role in migraine? Brain 128: 86–97, 2005.
- Andreou AP, Shields KG, Goadsby PJ. GABA and valproate modulate trigeminovascular nociceptive transmission in the thalamus. Neurobiol Dis 37: 314–323, 2010.
- Andreou AP, Goadsby PJ. Topiramate in the treatment of migraine: a kainate (glutamate) receptor antagonist within the trigeminothalamic pathway. Cephalalgia 31: 1343–1358, 2011.
- Afridi S, Giffin NJ, Kaube H, Friston KJ, Ward NS, Frackowiak RSJ, Goadsby PJ. A PET study in spontaneous migraine. Arch Neurol 62: 1270– 1275, 2005.
- Burstein R, Jakubowski M, Garcia-Nicas E, Kainz V, Bajwa Z, Hargreaves R, Becerra L, Borsook D. Thalamic sensitization transforms localized pain into widespread allodynia. Ann Neurol 68: 81–91, 2010.
- Giffin NJ, Ruggiero L, Lipton RB, Silberstein S, Tvedskov JF, Olesen J, Altman J, Goadsby PJ, Macrae A. Premonitory symptoms in migraine: an electronic diary study. Neurology 60: 935–940, 2003.
- Blau JN. Migraine prodromes separated from the aura: complete migraine. Br Med J 21: 658–660, 1980.
- Headache Classification Committee of the International Headache Society. The international classification of headache disorders, 3rd edition. Cephalalgia 33: 629–808, 2013.
- Lipton RB, Bigal ME, Ashina S, Burstein R, Silberstein S, Reed ML, Serrano D, Stewart WF. Cutaneous allodynia in the migraine population. Ann Neurol 63: 148–158, 2008.
- Gowers WR. A Manual of Diseases of the Nervous System. Philadelphia, PA: Blakiston, 1899, p. 1357.
- Burstein R, Collins B, Jakubowski M. Defeating migraine pain with triptans: a race against the development of cutaneous allodynia. Ann Neurol 55: 19–26, 2004.
- Bigal ME, Ashina S, Burstein R, Reed ML, Buse D, Serrano D, Lipton RB. Prevalence and characteristics of allodynia in headache sufferers: a population study. Neurology 70: 1525–1533, 2008.
- Stewart WF, Staffa J, Lipton RB, Ottman R. Familial risk of migraine: a population-based study. Ann Neurol 41: 166–172, 1997.
- Russell MB, Iselius L, Olesen J. Investigation of the inheritance of migraine by complex segregation analysis. Human Genet 96: 726–730, 1995.
- Lance JW, Goadsby PJ. Mechanism and Management of Headache. New York: Elsevier, 2005.
- Giffin NJ, Ruggiero L, Lipton RB, Silberstein S, Tvedskov JF, Olesen J, Altman J, Goadsby PJ, Macrae A. Premonitory symptoms in migraine: an electronic diary study. Neurology 60: 935–940, 2003.
- Maniyar FH, Sprenger T, Monteith T, Schankin C, Goadsby PJ. Brain activations in the premonitory phase of nitroglycerin triggered migraine attacks. Brain 137: 232–242, 2014.
- Nesbitt AD, Leschziner GD, Peatfield RC. Headache, drugs and sleep. Cephalalgia 34: 756–766, 2014.
- Barbanti P, Fabbrini G, Aurilia C, Vanacore N, Cruccu G. A case-control study on excessive daytime sleepiness in episodic migraine. Cephalalgia 27: 1115–1119, 2007.
- Blau JN. Sleep deprivation headache. Cephalalgia 10: 157–160, 1990.
- Mason P. Deconstructing endogenous pain modulations. J Neurophysiol 94: 1659–1663, 2005.
- Edvinsson L, Emson P, McCulloch J, Tatemoto K, Uddman R. Neuropeptide Y: cerebrovascular innervation and vasomotor effects in the cat. Neurosci Lett 43: 79–84, 1983.
- Eftekhari S, Salvatore CA, Johansson S, Chen TB, Zeng Z, Edvinsson L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res 1600: 93–109, 2015.
- Curran DA, Hinterberger H, Lance JW. Total plasma serotonin, 5-hydroxyindoleacetic acid and p-hydroxy-m-methoxymandelic acid excretion in normal and migrainous subjects. Brain 88: 997– 1010, 1965.
- Kimball RW, Friedman AP, Vallejo E. Effect of serotonin in migraine patients. Neurology 10: 107– 111, 1960.
- Lance JW, Anthony M, Hinterberger H. The control of cranial arteries by humoral mechanisms and its relation to the migraine syndrome. Headache 7: 93–102, 1967.
- Humphrey PPA, Feniuk W, Perren MJ, Beresford IJM, Skingle M, Whalley ET. Serotonin and migraine. Ann NY Acad Sci 600: 587–598, 1990.
- Humphrey PPA, Feniuk W, Perren MJ, Beresford IJM, Skingle M, Whalley ET. Serotonin and migraine. Ann NY Acad Sci 600: 587–598, 1990.
- Cohen ML, Johnson KW, Schenck KW, Phebus LA. Migraine therapy: relationship between serotonergic contractile receptors in canine and rabbit saphenous veins to human cerebral and coronary arteries. Cephalalgia 17: 631–638, 1997.
- Ullmer C, Schmuck K, Kalkman HO, Lubbert H. Expression of serotonin receptor mRNAs in blood vessels. FEBS Lett 370: 215–221, 1995.
- Dodick D, Lipton RB, Martin V, Papademetriou V, Rosamond W, MaassenVanDenBrink A, Loutfi H, Welch KM, Goadsby PJ, Hahn S, Hutchinson S, Matchar D, Silberstein S, Smith TR, Purdy RA, Saiers J. Consensus statement: cardiovascular safety profile of triptans (5-HT1B/1D Agonists) in the acute treatment of migraine. Headache 44: 414–425, 2004.
- Goadsby PJ. The pharmacology of headache. Prog Neurobiol 62: 509–525, 2000.
- Williamson DJ, Shepheard SL, Hill RG, Hargreaves RJ. The novel anti-migraine agent rizatriptan inhibits neurogenic dural vasodilation and extravasation. Eur J Pharmacol 328: 61–64, 1997.
- Mulderry PK, Ghatei MA, Spokes RA, Jones PM, Pierson AM, Hamid QA, Kanse S, Amara SG, Burrin JM, Legon S. Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience 25: 195–205, 1988.
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM. RAMPs regulate the transport and ligand specificity of the calcitoninreceptor-like receptor. Nature 393: 333–339, 1998.
- Recober A, Kuburas A, Zhang Z, Wemmie JA, Anderson MG, Russo AF. Role of calcitonin generelated peptide in light-aversive behavior: implications for migraine. J Neurosci 29: 8798–8804, 2009.
- Hay DL, Chen S, Lutz TA, Parkes DG, Roth JD. Amylin: pharmacology physiology and clinical potential. Pharmacol Rev 67: 564–600, 2015.
- Walker CS, Eftekhari S, Bower RL, Wilderman A, Insel PA, Edvinsson L, Waldvogel HJ, Jamaluddin MA, Russo AF, Hay DL. A second trigeminal CGRP receptor: function and expression of the AMY1 receptor. Ann Clin Transl Neurol 2: 595–608, 2015.
- Hokfelt T, Arvidsson U, Ceccatelli S, Cortes R, Cullheim S, Dagerlind A. Calcitonin gene-related peptide in the brain, spinal cord, and some peripheral systems. Ann NY Acad Sci 657: 119–134, 1992.
- Edvinsson L, Mulder H, Goadsby PJ, Uddman R. Calcitonin gene-related peptide and nitric oxide in the trigeminal ganglion: cerebral vasodilatation from trigeminal nerve stimulation involves mainly calcitonin gene-related peptide. J Auton Nerv Syst 70: 15–22, 1998.
- Edvinsson L, Eftekhari S, Salvatore CA, Warfvinge K. Cerebellar distribution of calcitonin gene-related peptide (CGRP) and its receptor components calcitonin receptor-like receptor (CLR) and receptor activity modifying protein 1 (RAMP1) in rat. Mol Cell Neurosci 46: 333–339, 2011.
- Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia 22: 54–61, 2002.
- Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28: 183–187, 1990.
- Gallai V, Sarchielli P, Floridi A, Franceschini M, Codini M, Trequattrini A, Palumbo R. Vasoactive peptides levels in the plasma of young migraine patients with and without aura assessed both interictally and ictally. Cephalalgia 15: 384–390, 1995.
- Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 33: 48–56, 1993.
- Storer RJ, Akerman S, Goadsby PJ. Calcitonin gene-related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br J Pharmacol 142: 1171–1181, 2004.
- Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nature Rev Neurol 6: 573–582, 2010.
- Connor KM, Shapiro RE, Diener HC, Lucas S, Kost J, Fan X, Fei K, Assaid C, Lines C, Ho TW. Randomized, controlled trial of telcagepant for the acute treatment of migraine. Neurology 73: 970–977, 2009.
- Connor KM, Aurora SK, Loeys T, Ashina M, Jones C, Giezek H, Massaad R, Williams-Diaz A, Lines C, Ho TW. Long-term tolerability of telcagepant for acute treatment of migraine in a randomized trial. Headache 51: 73–84, 2011.
- Ho TW, Ho AP, Chaitman BR, Johnson C, Mathew NT, Kost J, Fan X, Aurora SK, Brandes JL, Fei K, Beebe L, Lines C, Krucoff MW. Randomized, controlled study of telcagepant in patients with migraine and coronary artery disease. Headache 52: 224–235, 2012.
- Ho TW, Connor KM, Zhang Y, Pearlman E, Koppenhaver J, Fan X, Lines C, Edvinsson L, Goadsby PJ, Michelson D. Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology 83: 958–966, 2014.
- Marcus R, Goadsby PJ, Dodick D, Stock D, Manos G, Fischer TZ. BMS-927711 for the acute treatment of migraine: a double-blind, randomized, placebo controlled, dose-ranging trial. Cephalalgia 34: 114– 125, 2014.
- De Hoon J, Montieth D, Vermeersch S, Van Hecken A, Abu-Raddad E, Collins E, Schuetz T, Scherer J, Grayzel D. Safety pharmacokinetics, and pharmacodynamics of LY2951742: a monoclonal antibody targeting CGRP. Cephalalgia 33: 247–248, 2013.
- Silberstein, Stark, Lucas, Christie, Suzanne N., DeGryse, Turkel. PharmD; for the BoNTA-039 Study Group Botulinum Toxin Type A for the Prophylactic Treatment of Chronic Daily Headache: A Randomized, Double-Blind, Placebo-Controlled Trial. Mayo Clin Proc. 2005; 80:1126-37.
- Afridi SK, Giffin J, Kaube H, Frackowiak RS, Goadsby PJ. A PET study in spontaneous migraine. Arch Neurol 2005; 62: 1270-1275.
- Johnson KW, Bolay H. Neurogenic inflammatory mechanisms. In Olesen J, Goadsby PJ, Ramadan NM, Tfelt-Hansen P, Welch KWA eds. The Headaches, 3rd Ed. Philadelphia: Lippincott-Williams and Wilkins; 2006. Chapter 33; p. 309-319.
- Geppetti P, Rossi E, Chiarugi A, Benemei S. Antidromic vasodilation and the migraine mechanism. J Headache Pain 2012; 13:103-111.
- Palm-Meinders IH, Koppen H, Terwindt GM, et al. Structural brain changes in migraine. JAMA 2012; 308:1889-97.
- Iversen HK, Olesen J, Tfelt-Hansen P. Intravenous nitroglycerin as an experimental headache model Basic characteristics. Pain 38: 17–24, 1989.
- Akerman S, Williamson DJ, Kaube H, Goadsby PJ. Nitric oxide synthase inhibitors can antagonise neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br J Pharmacol 137: 62–68, 2002.
- Koulchitsky S, Fischer MJ, De Col R, Schlechtweg PM, Messlinger K. Biphasic response to nitric oxide of spinal trigeminal neurons with meningeal input in rat–possible implications for the pathophysiology of headaches. J Neurophysiol 92: 1320–1328, 2004.
- Lassen LH, Ashina M, Christiansen I, Ulrich V, Olesen J. Nitric oxide synthesis inhibition in migraine. Lancet 349: 401–402, 1997.
- Ahn AH. On the temporal relationship between throbbing migraine pain and arterial pulse. Headache 50: 1507–1510, 2010.
- Ray BS, Wolff HG. Experimental studies on headache. Pain sensitive structures of the head and their significance in headache. Arch Surg 41: 813– 856, 1940.
- Moskowitz MA. The neurobiology of vascular head pain. Ann Neurol 16: 157–168, 1984.
- Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 384: 560–563, 1996.
- Bernstein C, Burstein R. Sensitization of the trigeminovascular pathway: perspective and implications to migraine pathophysiology. J Clin Neurol 8: 89–99, 2012.
- Burstein R, Cutrer MF, Yarnitsky D. The development of cutaneous allodynia during a migraine attack. Brain 123: 1703–1709, 2000.
- Chronicle EP, Mulleners WM. Visual system dysfunction in migraine: a review of clinical and psychophysical findings. Cephalalgia 16: 525–535, 1996.
- Battelli L, Black KR, Wray SH. Transcranial magnetic stimulation of visual area V5 in migraine. Neurology 58: 1066–1069, 2002.
- Afra J. Intensity dependence of auditory evoked cortical potentials in migraine. Changes in the periictal period. Funct Neurol 20: 199–200, 2005.
- Boulloche N, Denuelle M, Payoux P, Fabre N, Trotter Y, Geraud G. Photophobia in migraine: an interictal PET study of cortical hyperexcitability and its modulation by pain. J Neurol Neurosurg Psychiatry 81: 978–984, 2010.
- Casucci, G., Villani, V., and Frediani, F. (2008). Central mechanism of action of antimigraine prophylactic drugs. Neurol. Sci. 29(Suppl. 1), S123– S126.
- Schoenen, J., Maertens de Noordhout, A., Timsit-Berthier, M., and Timsit, M. (1986). Contingent negative variation and efficacy of beta-blocking agents in migraine. Cephalalgia 6, 229–233.
- Dao W. Wang1,2, Akshitkumar M. Mistry1, Propranolol blocks cardiac and neuronal voltage-gated sodium channels. Pharmacology of Ion Channels and Channelopathies 31 December 2010 Sec https://doi.org/10.3389/fphar.2010.00144.
Patrocinante
